


QUALITY CONTROL OF MEDICINES
INTRODUCTION. The lack of national requirements regulating the quality of empty hard gelatin capsule shells (before filling with medicines) causes difficulties not only for capsule developers and manufacturers but also for experts evaluating regulatory submissions. This necessitates a comparative analysis of Russian and international requirements for the quality of hard gelatin capsule shells in order to draft a pharmacopoeia monograph on empty hard gelatin capsule shells.
AIM. This study aimed to analyse the level of quality requirements for hard gelatin capsule shells, the selection of quality attributes for testing, and the applicable methods and analytical procedures for the preparation of a draft monograph for the State Pharmacopoeia of the Russian Federation.
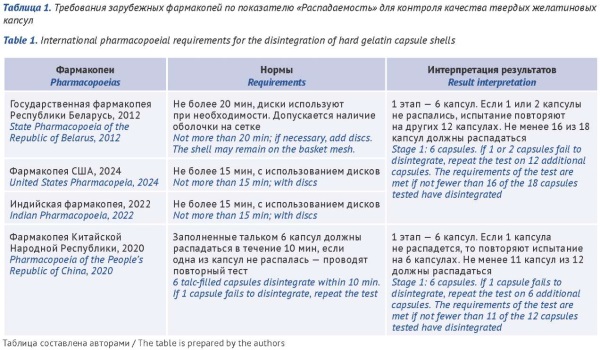
DISCUSSION. The results of this study indicate that the growing interest of manufacturers in producing capsule formulations is due to a number of features and advantages of this dosage form. Monographs on empty hard gelatin capsule shells are available in the State Pharmacopoeia of the Republic of Belarus, the Indian Pharmacopoeia, the Pharmacopoeia of the People’s Republic of China, the Korean
Pharmacopoeia, and the Japanese Pharmacopoeia. The pharmacopoeial requirements for hard gelatin capsule shells include testing for quality attributes, such as description, identification (gelatin, titanium dioxide, dyes, and preservatives), odour, average capsule mass, disintegration, loss on drying, microbiological quality, impurities (sulfate ash, heavy metals, and arsenic), and preservatives (parabens and sulfur dioxide). Compared with other pharmacopoeias, the pharmacopoeias of Belarus and China provide the most detailed approach to the control of hard gelatin capsule shells. According to the materials of registration dossiers for 65 medicinal products formulated as capsules, the hard gelatin capsule shells that are most frequently used in Russia are produced by eight manufacturers. The specifications of these manufacturers include all the common quality attributes described in the pharmacopoeias and additional tests for arsenic and lubricants.
CONCLUSIONS. The results of this comparative analysis of pharmacopoeial quality standards and manufacturers’ specifications for hard gelatin capsule shells justify the selection of the necessary quality attributes, limits, and requirements for inclusion in the draft monograph on empty hard gelatin capsule shells.
INTRODUCTION. Vegetable fatty oils can comprise over 50% of a medicinal product formulated as an oily solution (in some cases, 100%). The quality of the vegetable fatty oil used and the processes occurring in this oil have a significant impact on the quality of the medicinal product. The factors that influence the formation of impurities in vegetable fatty oils include the manufacturing process and storage and transportation conditions. Therefore, it is necessary to analyse national and international approaches to the assessment of impurities in vegetable fatty oils and medicinal products based on vegetable fatty oils.
AIM. This study aimed to analyse pharmacopoeial requirements for the control of impurities in vegetable fatty oils and summarise these requirements as recommendations for manufacturers of oily solution formulations containing 50–100% of vegetable fatty oils.
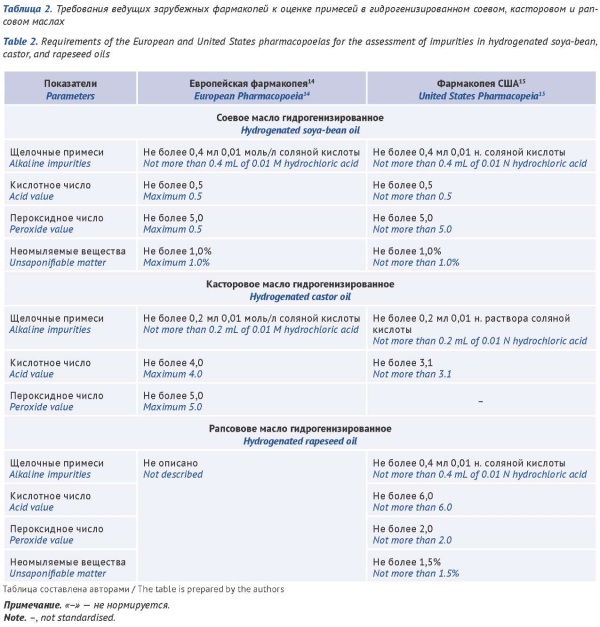
DISCUSSION. This study compared the requirements of national and international pharmacopoeias (State Pharmacopoeia of the Russian Federation, European Pharmacopoeia, and United States Pharmacopeia) for the control of impurities in vegetable fatty oils. The comparative analysis focused on the vegetable fatty oils most frequently used as solvents for the production of liquid dosage forms (sesame, olive, sunflower, soya-bean, rapeseed, and castor oils) as a case study. According to the results, as a rule, the impurity profiles provided by the monographs of the leading international pharmacopoeias for the same vegetable fatty oils differ, either qualitatively or quantitatively, and depend on the type of oil. Additionally, the pharmacopoeial limits for impurities in vegetable fatty oils vary depending on the intended dosage form. The State Pharmacopoeia of the Russian Federation lacks individual monographs for some of the vegetable fatty oils most frequently used in the production of medicinal products (sunflower, olive, sesame, and rapeseed oils). The State Pharmacopoeia of the Russian Federation requires that the quality of these oils should be assessed using the general approach described in the general monograph Vegetable fatty oils, whereas the requirements for the quality of medicinal products formulated as oily solutions are provided exclusively in the general monograph Solutions, which includes the acid value and peroxide value tests.
CONCLUSIONS. To draft pharmacopoeial monographs for oily solution formulations, it is necessary to standardise approaches to assessing impurities, in particular, to provide for mandatory acid value and peroxide value testing, as these attributes characterise the quality of oils during both production and storage.
INTRODUCTION. Antibiotic active substances have a distinct class of organic impurities other than related substances or organic solvents, namely, process-related impurities, such as N,N-dimethylaniline and 2-ethylhexanoic acid and its derivatives. These compounds are used as reagents in the production process, and national and international pharmacopoeias require that their residual levels should be controlled.
AIM. This study aimed to justify the requirements for the control of process-related impurities in antibiotic active substances.
MATERIALS AND METHODS. The study analysed general chapters and individual monographs on antibiotic active substances of the State Pharmacopoeia of the Russian Federation and international pharmacopoeias, including the European Pharmacopoeia (Ph. Eur.), the United States Pharmacopeia (USP), and The International Pharmacopoeia (Ph. Int.). The study also analysed registration dossiers for local and imported antibiotic active substances listed in the Russian State Register of Medicines. The study used comparative and content analysis methods.
RESULTS. The Ph. Eur. requires that 2-ethylhexanoic acid should be controlled in antibiotic active substances, such as semisynthetic and fermentation-derived antibiotics. In contrast, the USP does not include this quality attribute in individual monographs. The Ph. Eur. and USP individual monographs on semisynthetic antibiotics require manufacturers to control residual levels of N,N-dimethylaniline. This is probably due to the high toxicity of N,N-dimethylaniline. The relevant requirements of the State Pharmacopoeia of the Russian Federation are harmonised with those of the Ph. Eur. According to the analysis of registration dossiers, manufacturers replace 2-ethylhexanoic acid and N,N-dimethylaniline with less toxic reagents and, if necessary, control the residual levels of these alternative reagents in accordance with the general requirements for residual organic solvents.
CONCLUSIONS. Current technological processes can produce antibiotic active substances without the use of 2-ethylhexanoic acid and N,N-dimethylaniline. Therefore, the concept of process-related impurity control in active substances should be based on risk assessment. Furthermore, the relevant general chapters should require testing for 2-ethylhexanoic acid and N,N-dimethylaniline only if these compounds are used in the production process.
INTRODUCTION. An important quality attribute of medicines and excipients is the content of residual organic solvents (ROS). The general chapter on ROS (1.1.0008) of the State Pharmacopoeia of the Russian Federation (Ph. Rus.) does not provide analytical procedures for ROS identification, limit tests, and quantitative determination. This impedes the standardisation of approaches to the control of ROS.
AIM. This study aimed to analyse global pharmacopoeial practices for and methodological approaches to the control of ROS in order to prepare a draft general chapter on ROS.
DISCUSSION. According to the comparative analysis of the requirements for the control of ROS of the European Pharmacopoeia, the United States Pharmacopoeia, the Pharmacopoeia of the Eurasian Economic Union, and the Ph. Rus., the current Ph. Rus. general chapter lacks a description of chromatographic systems, and this description should be provided. The validation requirements for the relevant test procedures in neither of the analysed pharmacopoeias include an exhaustive list of equipment characteristics that are necessary and sufficient to satisfy all analytical conditions.
CONCLUSIONS. It is reasonable to harmonise the Ph. Rus. general chapter on ROS (1.1.0008) with global pharmacopoeial approaches to ROS determination by gas chromatography. The authors recommend supplementing the draft general chapter on ROS with analytical procedures for ROS identification, limit tests, and quantitative determination.
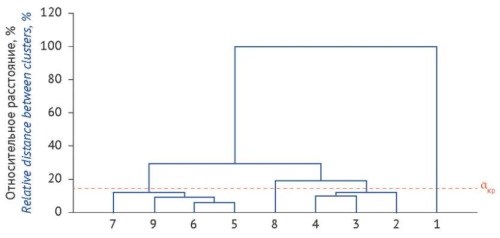
INTRODUCTION. Herbal drugs can often be difficult to identify and distinguish from the analysis of complex mixtures of herbal origin, it is reasonable to use additional identification methods, including hierarchical clustering of herbal drugs by their trace element content.
AIM. This study aimed to assess the possibility of using hierarchical clustering by trace element content for complex biological matrices (plant mixtures), with officinal chamomile species as a case study.
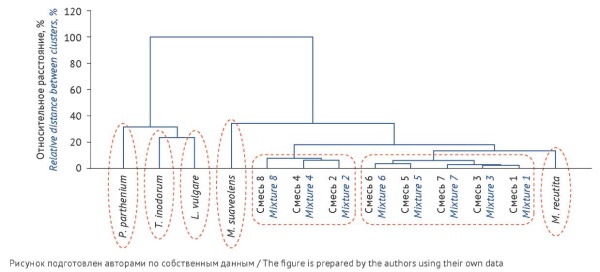
MATERIALS AND METHODS. Multiple morphologically similar species grow together with officinal chamomile species that are widely used in medical practice. The study examined the flowers and peduncles of Matricaria recutita L., Tanacetum parthenium (L.) Sch.Bip., Leucanthemum vulgare Lam., Tripleurospermum inodorum (L.) Sch.Bip., and Matricaria suaveolens Buchenau, as well as artificial mixtures of chamomile with adulterant species (as samples non-compliant with pharmacopoeial specifi ca - tions). The content of trace elements was determined by inductively coupled plasma mass spectrometry (ICP-MS). The statistical treatment of the results used Statistica 10 software with a built-in data analysis algorithm for multivariate experiments.
RESULTS. Having determined the content of 56 trace elements, the authors plotted a hierarchical cluster tree using cluster analysis methods. M. recutita and M. suaveolens were shown to be the closest to each other in terms of their trace element status, which correlated with their taxonomy. Artificial plant mixtures formed intermediate clusters that were observed between different chamomile species. Moreover, according to the criterion of dissimilarity, the trace element cluster of M. recutita significantly differed from the trace element clusters of all the adulterant species and mixtures containing more than 10% of the adulterant species.
CONCLUSIONS. This study demonstrated the possibility of using hierarchical clustering based on the content of trace elements in a biological object to analyse complex systems, including plant mixtures, and identify differences that may have discriminative value for the quality control of herbal drugs.
INTRODUCTION. Naftifine medicinal products are widely used in medical practice to treat fungal infections. A key quality attribute of a naftifine medicinal product is the content of the active substance. It is essential to develop quantitative analytical procedures for the routine control of medicines with a particular focus on reducing time and costs.
AIM. This study aimed to update the analytical procedures for the quantitative determination of naftifine and its impurities in medicines by high-performance liquid chromatography (HPLC) with small-volume columns providing a reduction in assay time and reagent consumption.
MATERIALS AND METHODS. This study focused on the active substance naftifine and naftifine-based medicinal products, including a 1% naftifine alcohol solution and a 1% naftifine cream for cutaneous use. The solutions were analysed on Agilent 1200 Infinity and Agilent Infinity II 1290 liquid chromatography systems equipped with diode-array detectors and several chromatographic columns: XBridge Phenyl, 20×4.6 mm, 2.5 μm; XBridge Phenyl, 20×4.6 mm, 3.5 μm; and Acquity BEH Phenyl, 75×2.1 mm, 1.7 μm. The specificity of the analytical procedure was evaluated using spiking solutions of N-methyl-1-naphthalenemethylamine and cinnamaldehyde as well as naftifine solutions after chemical, thermal, and photolytic decomposition.
RESULTS. The authors identified the optimal non-toxic solvent (0.1% orthophosphoric acid solution) and demonstrated the applicability of different solvents to different dosage forms. Additionally, the selected analytical conditions included the following: 10 μg/mL naftifine solutions were chromatographed on an XBridge Phenyl column (20×4.6 mm; 2.5 μm) using a gradient of 0.1% perchloric acid and acetonitrile at an elution rate of 1 mL/min. The study showed that the selected detection wavelength of 254 nm provided the best signal-to-noise ratio for the naftifine peak. The reproducibility of the developed quantitative determination procedure was confirmed by validation in accordance with the current requirements of the State Pharmacopoeia of the Russian Federation. The specificity of the analytical procedure was shown by chromatographic analysis of the solvent, mobile phase, and solutions containing the main naftifine impurities. The validation study confirmed the linearity of the analytical procedure in the range of 80–120% (with a correlation coefficient of 0.995). During the accuracy validation, the recovery rate was 100.2%. The validation study demonstrated the robustness of the analytical procedure to minor changes in the chromatographic parameters. The naftifine retention time amounted to approximately 2 minutes.
CONCLUSIONS. The authors developed a selective and sensitive HPLC-based analytical procedure for the quantitative determination of naftifine in medicines. This analytical procedure provides for a reduction in assay time and reagent consumption, and its compliance with the validation acceptance criteria indicates its suitability and reproducibility.
HERBAL MEDICINAL PRODUCTS
INTRODUCTION. Ganoderma spp. have been used as a traditional oriental medicine and a bioactive dietary supplement. These fungi are a promising source of effective antioxidants. Currently, there is no regulatory framework to control the quality of this herbal drug and its bioactive components in the Russian Federation and the Republic of Belarus. Therefore, it is essential to study the chemical composition and pharmacological activity spectrum of G. lingzhi and G. lucidum extracts.
AIM. The aim of this study was to determine the chemical composition and antioxidant activity of G. lingzhi and G. lucidum fruiting body extracts.
MATERIALS AND METHODS. The study focused on pure cultures of G. lingzhi and G. lucidum obtained from the fungal species collection of the Forest Institute of the National Academy of Sciences of Belarus. Fungal biomass was grown using two substrates, including alder sawdust (1–3 mm fraction) and oak shavings (5–10 mm fraction). The fungal biomass was extracted using repeated maceration with 70% ethanol. The study tested the free radical-scavenging activity of the extracts in reactions with the stable free radical 2,2-diphenyl-1-picrylhydrazyl (DPPH) and the radical cation derived from 2,2’-azinobis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS). The chemical composition was analysed by high-performance liquid chromatography–mass spectrometry (HPLC–MS). The assays for phenols, steroids, and triterpenes used spectrophotometry.
RESULTS. The extract of G. lucidum strain 334 cultivated on the alder substrate demonstrated the highest free radical-scavenging activity (IC50=3.1±0.2 μg/mL (DPPH), IC50=3.7±0.2 μg/mL (ABTS)), the highest phenolic content (326.2±16.5 µmol/g), and the highest triterpene content (2.00±0.11 mmol/g) of all the studied extracts. The antioxidant activity of the extracts of G. lingzhi and G. lucidum may be attributed to the content of ganoderic acid D, lucidenic acid D, naringenin, and other phenolic compounds.
CONCLUSION. The high yield of extracts with a significant radical-scavenging activity makes artificially cultivated G. lingzhi and G. lucidum mushrooms a promising source of natural antioxidants.
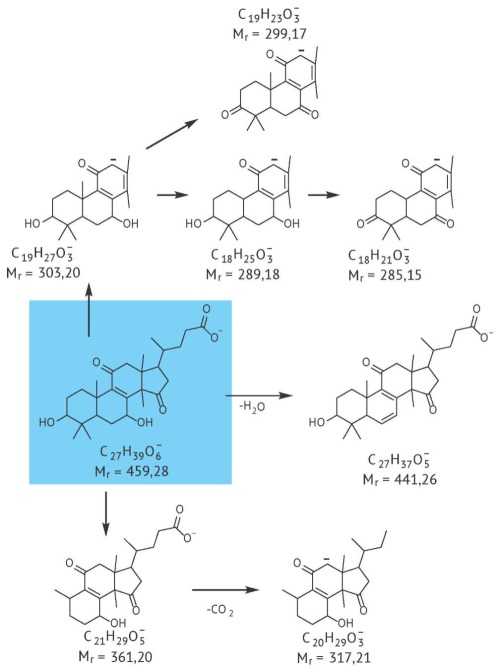
INTRODUCTION. The compendial analytical procedure for determining the content of heavy metals and arsenic impurities in herbal drugs does not differentiate between endogenous and exogenous elemental impurities. Higher-precision data on the microelement composition of plants, particularly data on endogenous and exogenous impurities, could be instrumental in chemosystematics and in the identification of raw materials.
AIM. This study aimed to modify the analytical procedure for quantifying macro- and microelements in herbal drugs with the intention to eliminate the influence of exogenous pollutants on test results.
MATERIALS AND METHODS. The study focused on herbal drugs from the Boraginaceae family, including reproductive shoots of lungwort (Pulmonaria mollis Wulf. ex Hornem) and leaves of common borage (Borago officinalis L.). A portion was spiked with elemental pollutants. All herbal drugs were ground and fractionated by sieving. Quantitative determination was performed by inductively coupled plasma mass spectrometry. Statistical analysis of the results used Microsoft Excel and Statistica 8 software.
RESULTS. A total of 30 elements were quantified in 9 fractions of herbal drugs with different particle sizes. Cluster analysis showed that unfractionated herbal drugs and the fraction comprising particles smaller than 0.2 mm formed sepa rate clusters, indicating significant differences in elemental compositions. Grubbs’ test confirmed that these two samples were significantly different from the others. The fraction of particles smaller than 0.2 mm had the maximum content of the spiked elements; more than 90% of the spiked amounts were recovered from this fraction. The high content of elements observed in unfractionated herbal drugs was due to the plant parts that would have passed the 0.2 mm sieve had they been ground and fractionated.
CONCLUSIONS. The use of unfractionated herbal drugs leads to a systematic error in the results of elemental content determination, which significantly hinders the applicability of such results for the identification of herbal drugs. To ensure reliable results in the determination of the microelement status of plants, the authors suggest performing additional fractionation of herbal drugs and discarding the fraction under 0.2 mm.
PHARMACEUTICAL MANUFACTURING
INTRODUCTION. The pharmaceutical industry aims at developing sustainable resource-saving technologies for manufacturing medicines. Approaches to the effective use of herbal drugs include applying technologies yielding compounds with various pharmacological effects in one production cycle. To date, no standardised herbal drug processing technology has been designed to produce calendula flower-based medicinal products with various pharmacological effects. The Member States of the Eurasian Economic Union (EAEU) process calendula flowers by extraction methods yielding one fraction of biologically active substances per cycle (either only flavonoids or only carotenoids).
AIM. This article aimed to investigate the possibility of stepwise processing of calendula flowers to obtain biologically active substances with different polarities (carotenoids, flavonoids, and polysaccharides) per cycle.
MATERIALS AND METHODS. The authors extracted carotenoids using hexane. The study included additional thermal pretreatment of the herbal drug. Carotenoids and flavonoids were quantified by spectrophotometry, and polysaccharides were quantified by gravimetric analysis. After hexane removal, the dry oily residue was dissolved in oil with mechanical stirring to obtain the oil extract. The study included a three-step processing method that comprised hexane extraction (extraction of carotenoids with a low-polarity organic solvent), extraction with a water–organic solvent system (extraction of flavonoids), and aqueous extraction (precipitation of polysaccharides).
RESULTS. The lipophilic extracts obtained by single hexane extraction or by single hexane extraction and heat treatment had the maximum carotenoid content (~4%), which was 10 times the carotenoid content in the extracts obtained using a water–organic solvent system. Oil extracts were prepared from the dry residue left after removing hexane by distillation at its boiling point. The content of biologically active substances in the oil extracts was comparable to that in the intact herbal drug. Water–organic solvent extraction yielded 68.5% more flavonoids from the herbal drug extracted with hexane than from the intact herbal drug. In contrast, aqueous extraction applied to the intact herbal drug yielded 1.7 times the amount of flavonoids obtained by aqueous extraction preceded by hexane extraction. The water-soluble polysaccharide fraction extracted from the intact herbal drug had 10 times the flavonoid content observed in the fraction extracted from the processed herbal drug. Each subsequent processing step decreased the total polysaccharide content and the content of individual polysaccharide fractions and increased their purity. The ratio of polysaccharide fractions remained unchanged across all processing methods tested.
CONCLUSIONS. Stepwise processing of calendula flowers ensured that one processing cycle provided three fractions containing increased amounts of carotenoids, flavonoids, and polysaccharides. Relative to the intact herbal drug, there was a 3.3-fold increase in the yield of carotenoids, and the yields of flavonoids and polysaccharides increased by 44.8 and 41.3%, respectively. The resulting product had a higher degree of purity than the products obtained by pretreatment and double hexane extraction or by using the intact herbal drug. The stepwise processing method is advisable for the production of medicines that are enriched with a specific group of biologically active substances or contain smaller amounts of impurities.
PRECLINICAL STUDIES

INTRODUCTION. Stem cell therapy is a promising treatment method for various diseases and injuries, but its safety has yet to be determined. Therefore, studying the safety of administering a xenogeneic cell-based medicinal product (CBMP) into the retro-orbital venous sinus is essential for developing protocols for further studies of potential medicinal products for neurological conditions.
AIM. The aim of the study was to determine the optimal dose of a CBMP derived from glial progenitor cells (GPCs) and to evaluate its safety during retrobulbar administration in C57BL/6J mice.
MATERIALS AND METHODS. GPCs were derived from human induced pluripotent stem cells by stepwise differentiation and cultured in DMEM/F12 supplemented with epidermal growth factor and ciliary neurotrophic factor. Matrigel was used as a substrate. GPCs were injected into the retro-orbital venous sinus of male C57BL/6J mice under isoflurane anaesthesia once a week for two months. The study analysed changes in biochemical blood parameters and behaviour. The quantities of activated astrocytes and glial cells were determined by postmortem immunohistochemical staining.
RESULTS. The administration of GPCs at a dose of 500×103 cells/mouse, which was selected using literature data, induced an increase in the plasma levels of ala nine aminotransferase and aspartate aminotransferase. This could indicate cell damage and the development of inflammatory reactions. At doses reduced to one-third the initial GPC concentration or lower, the biochemical blood parameters of the treatment groups did not differ significantly from those of the control group. There were no significant differences in neuroinflammatory markers between the groups receiving GPCs at different doses, except for an increase in astrocyte activation at a dose of 150×103 cells/mouse, which could potentially indicate inflammatory processes in the brain. The study detected no pathological changes in the brain or cell damage markers in the blood of mice after retrobulbar GPC injections of 15×103 or 50×103 cells/mouse.
CONCLUSIONS. The study results indicate that long-term therapy with GPCs is potentially safe for mice if the dose is optimal. The authors suggest using the optimal doses and the administration route established in this study for further research into the safety of intravenous administration of CBMPs for neurological conditions.

ISSN 3034-3453 (Online)





























