


AUTHORITATIVE OPINION

The ultimate goal of preclinical studies is to generate data that can be used to make conclusions about the potential effects of a new medicine on the human body and to properly proceed to clinical trials. In this interview, Marina N. Makarova, Doctor of Medical Sciences, Director of the RMC “HOME OF PHARMACY”, shares her views on conducting preclinical studies in Russia.
REGULATORY PRACTICE AND RECOMMENDATIONS
INTRODUCTION. Currently in the Russian Federation and the Eurasian Economic Union (EAEU), the conduct of non-clinical studies of radiopharmaceuticals is regulated by general guidelines on non-clinical studies, which are applicable to multiple groups of medicinal products but do not accommodate the characteristics of radiopharmaceuticals.
AIM. The study aimed to substantiate the need for methodological guidelines regulating non-clinical studies of radiopharmaceuticals, taking into account global regulatory practice.
DISCUSSION. The authors analysed the Russian and EAEU guidelines regulating non-clinical studies in general as well as international publications on non-clinical studies of radiopharmaceuticals, including guidelines by the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the International Atomic Energy Agency (IAEA). The analysis revealed the need for terminology harmonisation. This article describes the aspects specific to radiopharmaceuticals that limit the applicability of general guidelines to non-clinical studies of radiopharmaceuticals. Subchronic toxicity studies based on inactive formulations do not account for the contribution of radionuclide emission to the biological effects of therapeutic radiopharmaceuticals. The authors suggest that systemic toxicity studies of finished dosage forms should use single-dose chronic toxicity protocols.
CONCLUSIONS. It is necessary to develop and implement up-to-date methodological guidelines for conducting non-clinical studies of radiopharmaceuticals to reduce potential risks in the application of these medicinal products. This requires factoring in different requirements for diagnostic and therapeutic radiopharmaceuticals, with due consideration of radiation safety requirements. When drafting guidelines for non-clinical studies of radiopharmaceuticals, regulators should keep in mind the applicable guidelines by the FDA, EMA, and IAEA. A list of mandatory studies should be compiled, and uninformative studies should be excluded from the requirements for radiopharmaceuticals.
SAFETY OF MEDICINES
INTRODUCTION. In the current practice of preclinical safety studies of pharmacologically active substances, standard neurotoxicity assessment procedures are mainly aimed at diagnosing higher nervous activity and behavioural disorders. However, it is the structures of the peripheral nervous system that are particularly susceptible to drug-induced neurotoxicity, which renders these structures an easy target and leads to a high incidence of neurotoxic side effects. These circumstances dictate the importance of refining methodological approaches to the assessment of toxic injury in the peripheral nervous system.
AIM. The study aimed to analyse the current methodological level of clinical and functional tests for assessing the toxic effects of pharmacologically active substances on the structures of the peripheral nervous system, as well as to formulate practical recommendations for using these tests in preclinical studies in rodents.
DISCUSSION. Rodents are considered the optimal test system for preclinical studies of pharmacologically active substances, but it is impossible to reproduce the entire neurological examination that is conducted to identify clinical equivalents of neurotoxicity in humans using these animals. This article presents a systematic approach to using available diagnostic tests to increase the translatability of data. The article briefly describes the neurological deficits due to adverse drug reactions in humans, as well as the main toxidromes that can also occur in animals. Based on a literature review and experience, the authors provide practical recommendations for performing basic tests to study the strength and tone of muscles, the state of physiological reflexes, the coordination of movements, and various types of sensitivities in rodents. The article provides a brief overview of the diagnostic utility of electrophysiological testing for identifying toxic damage to the peripheral nervous system. The following tests are recommended as a minimum list of primary screening techniques for detecting neurotoxic side effects in study animals: a resting posture assessment, the beam walking test, the horizontal bar test, the digit abduction score assay, the tail flick test, and the Preyer reflex test.
CONCLUSIONS. The results of a comprehensive assessment of neurological deficits in rodent experiments should be analysed from a clinically relevant perspective— that is, with a focus on topical diagnosis and common pathological process components. It is advisable to verify the pathological process at the level of the peripheral nervous system using a set of electrophysiological techniques.
INTRODUCTION. Functional examination of the urinary system, and particularly the kidneys, is an important challenge in preclinical studies. Currently, there is no generally recognised and detailed approach to drug-induced nephrotoxicity detection in vivo, nor are there clear criteria for its assessment.
AIM. This study aimed to analyse and systematise instrumental and laboratory methods for the assessment of urinary system function in laboratory animals and to identify the basic principles for studying drug-induced nephrotoxic effects.
DISCUSSION. The study analysed the advantages and limitations of the methods used to study the nephrotoxicity of medicinal products, with considerations for the use of these methods in small and large laboratory animals. The effects of a test substance on the urinary system should first be evaluated using minimally invasive methods. One of these methods is urinalysis. For urinalysis, important considerations include the sampling technique, the volume of the biomaterial, and the turnaround time between urine collection and analysis. Ultrasonography is the most accessible instrumental method in preclinical studies. Ultrasonography can assess organ position, size, structure, and echogenicity and detect abnormalities and changes in real time. Dif ferent method settings are preferred for each species of laboratory animal. Further analysis can include macroscopic examination of organs, measurement of their masses, and microscopic analysis of tissues. Visual assessment should cover the size, colour, and consistency of the ureters, bladder, and kidneys. Nephrotoxicity may manifest as increased apoptosis, vacuolation of renal tubular epithelial cells, epithelial degeneration or dystrophy, oedema, diapedesis-associated haemorrhages, acute tubular and papillary necrosis, necrosis of the Bowman–Schumlansky capsule, casts and crystals in the tubular lumen, glomerulopathy with the corresponding changes, and inflammatory and vascular reactions.
CONCLUSIONS. The study analysed and systematised instrumental and laboratory methods for assessing the functional state of the urinary system in preclinical studies. The authors outlined the basic principles for a structured and comprehensive study of the potential nephrotoxicity of novel medicines. The assessment of nephrotoxicity should start with simple and minimally invasive laboratory and instrumental methods, which include general urinalysis and microscopic examination of urine sediment. These methods can detect organ dysfunction that has not yet presented with an associated anatomical lesion. A more in-depth analysis should involve histological and immunohistochemical methods to examine the urinary tissues of laboratory animals.
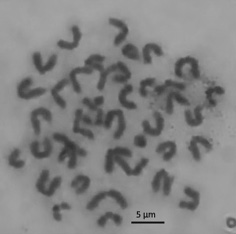
INTRODUCTION. Metaphase analysis is used to assess the mutagenicity of medicines, and its accuracy depends directly on the quality of cytogenetic preparations. Existing procedures for cytogenetic sample preparation require optimisation because of their time and labour intensiveness and potential for artefactual chromosome loss that leads to errors in the interpretation of analytical results.
AIM. This study aimed to optimise a cytogenetic sample preparation procedure for mammalian bone marrow cells for in vivo metaphase analysis.
MATERIALS AND METHODS. The study was performed in randomly bred male mice weighing 18–20 g. The authors prepared cytogenetic samples of bone marrow cells according to the procedures set forth in the Guidelines for Conducting Preclinical Studies of Drugs (A.N. Mironov (ed.), 2012), and GOST 34659-2020, Methods for Testing the Impact of Chemical Products on the Human Body. Assessment of Chromosomal Aberrations in Bone Marrow Cells of Mammals, as described and with modifications. Sample preparation involved using Hanks’ solution, sodium citrate and potassium chloride solutions, acetic acid, methyl alcohol, medium 199, HEPES, and Giemsa stain.
RESULTS. The authors conducted an experimental comparison of the characteristics of cytogenetic preparations of mammalian bone marrow cells for in vivo metaphase analysis prepared using several methods. The samples prepared in accordance with the Guidelines for Conducting Preclinical Studies of Drugs exhibited the highest percentage of analysable metaphase plates, in comparison with those obtained using the other sample preparation procedures. The authors optimised the sample prepara tion conditions, including the composition of the hypotonic solution (0.56% KCl), the time of hypotonic treatment (20 min), the time of fixation (12 h), and the composition of the culture medium (liquid medium 199 with Hanks’ salts and glutamine). The authors introduced a step of preliminary chromosome fixation with 6% acetic acid, which generally contributed to an increase in the proportion of analysable metaphase plates by reducing the scattering and sticking of chromosomes.
CONCLUSIONS. The authors presented a modified sample preparation procedure for cytogenetic preparations that, compared with the previous procedures, offers an increase in the percentage of analysable metaphase plates to 56%, a 12% rise in the percentage of full sets of chromosomes (n=40), and a 2–10-hour reduction in the preparation time.
BIOANALYTICAL METHODS
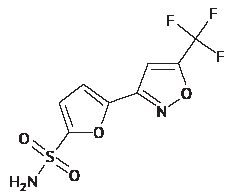
INTRODUCTION. Systemic exposure studies of a selective carbonic anhydrase II inhibitor, the isoxazole derivative 5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (TFISA), require evaluating its pharmacokinetics in whole blood because the compound can accumulate in erythrocytes. Currently, no bioanalytical procedures have been developed to achieve this.
AIM. This study aimed to develop a bioanalytical procedure for the determination of TFISA and its metabolites (N-hydroxy-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide and N-acetyl-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide) in the blood of laboratory animals and compare the pharmacokinetics of TFISA ophthalmic suspension in rats after a single ocular or intraperitoneal administration.
MATERIALS AND METHODS. The quantitative determination was performed by high-performance liquid chromatography–tandem mass spectrometry (HPLC-MS/MS) using rat and rabbit blood samples. The chromatographic separation used a Zorbax Eclipse Plus C18 column (150×3.0 mm, 3.5 µm) and a gradient elution system of 0.1% aqueous formic acid and methanol. The multiple reaction monitoring mass spectrometry mode was used for detection. The pharmacokinetics study was conducted in 2 groups of 6 Wistar rats (3 males and 3 females per group). Group 1 received an instillation of 1% TFISA ophthalmic suspension in each eye at a dose of 3.7 mg/kg. Group 2 received an intraperitoneal injection of the same product at the same dose. Blood samples were collected at baseline and at several intervals after administration.
RESULTS. The authors developed a bioanalytical procedure for the determination of TFISA and its metabolites in the blood of laboratory animals (rabbits and rats). This HPLC-MS/MS procedure was fully validated in accordance with the requirements of the EAEU legislation and the ICH M10 guideline. The analytical ranges in blood were 20–20000 for TFISA, 2–2000 for the N-hydroxy metabolite, and 0.1–100.0 ng/mL for the N-acetyl metabolite. The maximum blood levels after ocular instillation (mean±SD) were 8173±1491 for TFISA, 694±271 for the N-hydroxy metabolite, and 6.33±1.51 ng/mL for the N-acetyl metabolite. The half-lives for this route of administration were 58±10 (TFISA), 70±24 (N-hydroxy metabolite), and 14±3 h (N-acetyl metabolite). The bioavailability of TFISA was 90.18%. CONCLUSIONS. The developed and validated bioanalytical procedure for the determination of TFISA and its metabolites in the blood of laboratory animals has been successfully applied to samples of rat whole blood. According to the study of ophthalmic suspension pharmacokinetics, TFISA and its metabolites have long halflives and high bioavailability.
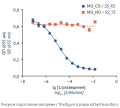
INTRODUCTION. Bioanalytical techniques are characterised by greater variability and lower stability than physicochemical methods because live test systems are inherently labile. Since regulatory standards do not establish a unified approach, the selection of system suitability criteria and acceptance criteria for test results is based on validation studies.
AIM. This study aimed to validate an analytical procedure for evaluating the biological activity of a medicinal product based on the investigational tocilizumab biosimilar GNR-087 and determine the quantitative limits for system suitability criteria and acceptance criteria for test results.
MATERIALS AND METHODS. The biological activity of the investigational tocilizumab biosimilar was assessed by the inhibition of IL-6-induced secreted embryonic alkaline phosphatase (SEAP) expression by HEK-Blue™ IL-6 cells. Statistical processing of the obtained results was performed using Prism 6.0 software.
RESULTS. The specificity of the analytical procedure was confirmed by the dose-dependent inhibition of IL-6-induced SEAP expression by cells observed with tocilizumab. The analytical procedure was linear, with a coefficient of determination R2≥ 0.99. The precision of the analytical procedure was satisfactory; its repeat ability varied from 2 to 9%, and its intermediate precision was 14%. The recovery coefficients (Rc) for spiked samples simulating activity levels of 60–140%, including blinded samples, ranged from 80 to 120%. The theoretical values of relative potency (RP) were within the confidence intervals of the mean relative potency estimates, which confirmed the accuracy of the analytical procedure. The validation confirmed the robustness of the analytical procedure to controlled variations, including the use of reporter cells at different passages (with a coefficient of variation for relative potency estimates (CVRP) of 10%), different IL-6 lots (with a CVRP of 1%), and different SEAP detection reagent lots (with a CVRP of 3%); the Rc remained in the range of 80–120% of the nominal RP value.
CONCLUSIONS. The analytical procedure for evaluating the biological activity of the investigational tocilizumab biosimilar meets the validation criteria, including accuracy, linearity, precision, specificity, and robustness. The study established system suitability criteria and acceptable limits for biological assay results. This analytical procedure can be used both for routine biological activity control and for demonstrating the biosimilarity of new medicinal products to the original (reference) tocilizumab-based medicinal product.

INTRODUCTION. The tendency towards reducing the use of laboratory animals in pharmaceutical quality assessment, along with the development of new technologies for gonadotrophin production, necessitate the development of novel assays to determine the biological activity of natural and recombinant anterior pituitary preparations.
AIM. This study aimed to select the optimum conditions for the determination of the biological activity of luteinising hormone (LH) in urinary and genetically engineered LH preparations by tests in randombred and Sprague Dawley rats.
MATERIALS AND METHODS. The biological activity of urinary and recombinant LH preparations was determined in vivo in a three-dose randomised Steelman–Pohley bioassay, which compared the biological activity of the test samples to a reference standard (RS). The analysis used two years’ worth of test results. The RS used was the WHO international standard containing 183 IU of follicle stimulating hormone bioactivity and 177 IU of LH bioactivity per ampoule. The bioassay was performed in immature male randombred and Sprague Dawley rats; the bioassay conditions were selected according to the rat type.
RESULTS. The results of the in vivo determination of LH biological activity in male rats were compared by the standard deviation to slope ratio (s/b), which helps estimate response variation and dose dependence at the same time. The study demonstrated the possibility of using Sprague Dawley rats (s/b=0.82±0.44) for the determination of LH biological activity along with randombred animals (s/b=0.68±0.58). The s/b ratios were close, although Sprague Dawley rats and randombred rats were used in the assay at a 1:2 ratio.
CONCLUSIONS. It is possible to determine the biological activity of LH from menopausal gonadotrophin preparations in both randombred and Sprague Dawley male rats. The use of Sprague Dawley rats for testing can confirm the reliability of test results using fewer animals than needed for standard tests; this is not only humane but also cost effective.
CLINICAL STUDIES
INTRODUCTION. A well-planned design of a clinical trial (CT) ensures valid results in assessing the efficacy and safety of medicines for human use. However, at present, there are no clear criteria for selecting the basic elements underlying the development of a CT design. This lack of selection criteria primarily concerns planning research hypotheses, calculating the expected therapeutic effect, statistical significance level, and study power, and selecting statistical models for sample size calculation.
AIM. The authors aimed to systematise and harmonise the technical requirements for sample size determination in designing CTs.
DISCUSSION. First, this article describes the basic requirements for and methodological approaches to designing CTs to assess the efficacy of medicines and to confirm their safety. Next, the article presents the basic principles for calculating the sample size to ensure the required CT power. Finally, the article covers the mathematical models describing the null and alternative hypotheses used in the development of basic statistical designs for efficacy and safety studies. A general requirement for the quality of a study sample is to ensure its representativeness, that is, its compliance with the target CT population. The selection of a mathematical (probabilistic) model to formulate research hypotheses and calculate study samples representative of the target population is based on general data from systematic reviews of previous studies on the therapeutic effects of the study product and the specific characteristics of the target population. In addition, model selection relies on the classification of the study product. Sample size calculation requires defining and justifying certain criteria at the stage of CT design and statistical model development, in line with the general requirements for representativeness. Software for calculating the statistical power and required sample size facilitates routine CT planning.
CONCLUSIONS. The sample size determination requires more than the application of basic statistical models. Given the multitude of CT designs and methodological approaches to CT planning, treatment regimens, and data collection and analysis, it is necessary to consider the statistical design of each CT on a case-by-case basis. This consideration should include assessments of individual cases, survival analysis methods, relative risks, diagnostic tests, and adaptive and other infrequent CT designs. The above highlights the need to develop additional guidelines and information resources that would explain and demonstrate the use of probabilistic statistics. The resulting national standards should be harmonised with international standards.
INTRODUCTION. Generalised anxiety disorder (GAD) is the least studied anxiety disorder, as patients present with comorbid mood disorders. Finding effective treatment methods for GAD is of the utmost importance; therefore, it is essential to develop novel medicinal products for GAD. Proper clinical programme design is key to obtaining reliable data on the effectiveness and safety of a medicinal product. Currently, the Russian Federation lacks methodological guidelines for clinical studies of these medcinal products, and there is a need for developing such guidelines.
AIM. This review aimed to assess the possibility of applying the methodological approaches described in international guidelines to Russian clinical trials to develop medicinal products for GAD.
DISCUSSION. Having analysed the main provisions of the Guideline on the clinical investigation of medicinal products indicated for generalised anxiety disorder by the European Medicines Agency (EMA), the authors of this review outlined the main stages of clinical development and the methodology for using clinical data to evaluate the safety and efficacy of medicinal products for GAD. Clinical development programmes for these medicinal products should take into account research staging and mandatory long-term safety and additive effect assessments. This review highlights aspects of selecting the design, population, and primary and secondary endpoints for a clinical trial. Particular attention is paid to the consideration of comorbidities in patients.
CONCLUSION. The provisions set forth in the EMA guideline can inform the development of national guidelines for studying medicinal products for GAD.
MICROBIOLOGICAL TESTING
INTRODUCTION. Microbiological testing of any medicine begins with the determination of its antimicrobial activity—that is, its effect on the growth of aerobic bacteria, yeasts, and moulds. Therefore, the improvement of microbiological testing methods to enhance the quality of resulting data remains a high priority.
AIM. The study aimed to develop a methodological approach to testing non-sterile medicines with antimicrobial effects for microbiological quality, as well as to improve the accuracy and reliability of existing microbiological testing methods.
MATERIALS AND METHODS. The study used 12 non-sterile pharmaceuticals, 5 diluents (phosphate buffer solution, phosphate buffer solution with polysorbate 80 (1% and 5%), neutralising fluid, and trypticase soy broth), culture media, and test strains required for microbiological quality testing according to the State Pharmacopoeia of the Russian Federation and the Pharmacopoeia of the Eurasian Economic Union.
RESULTS. This study established microbiological testing conditions for non-sterile medicines with known antimicrobial effects. The analytical procedures developed in the study were tested on 10 medicines. The authors demonstrated the applicability of analytical procedures for the quantitative determination of microscopic fungi in samples with fungistatic activity. The proliferation factors in growth promotion tests with Candida albicans and Aspergillus brasiliensis were within the acceptable range of 76–128% of the calculated value for a standardised inoculum. The authors proposed a modification to the analytical procedure for the qualitative determination of Escherichia coli; the modification involved using a 50 mL sample at a 1:50 dilution, corresponding to 1 g of the test product, for transfer to 450 mL of an appropriate culture medium. Having compared diluents containing non-specific agents neutralising antimicrobial activity, the authors showed that the neutralising fluid should be preferred.
CONCLUSIONS. The authors proposed a methodological approach to microbiological quality testing of medicines, postulating that the results should be considered reliable only if they account for the antimicrobial activity of the test product. The study demonstrated the feasibility of increasing the sample volume up to 10 mL and using a dilution exhibiting no antimicrobial activity (e.g., 1:100). The authors recommended using 150 mm Petri dishes, 50 mL of an agarised culture medium, and diluents with up to 5% of polysorbate 80 for plating. The study demonstrated the suitability of using a dilution exhibiting no antimicrobial activity. The study results indicated the need to increase the sample volume to be proportionally transferred to a culture medium; the sample should contain the amount of the test product regulated by pharmacopoeias (i.e., not less than 1 g (1 mL)). Diluents with up to 5% of polysorbate 80 were recommended for neutralising bacteriostatic and fungistatic effects.
EXPERT OPINION
This article provides recommendations for documenting an analytical procedure for water determination by Karl Fischer titration in a product specification file (PSF). The article contains a description of specific aspects associated with the test and a template for the typical content and layout of the corresponding PSF section. A unified approach to the PSF content and layout will ensure error-free testing, reliable results, and streamlined regulatory assessments.

ISSN 3034-3453 (Online)





























