


MAIN TOPIC: HARMONISATION OF PHARMACOPOEIAL STANDARDS: EXPERIENCE OF THE RUSSIAN FEDERATION

This interview examines key aspects of harmonizing pharmacopoeial standards for medicines in the context of a globalized pharmaceutical market. The author analyzes the experience of international organizations (WHO, ICH, PDG) and assesses the feasibility of creating uniform global quality standards. Particular attention is paid to the development of the EAEU Pharmacopoeia, the principles of terminology unification, the development of the Nomenclature of Dosage Forms, and the methodological differences between leading pharmacopoeias (USP, Ph. Eur., BP, JP). The main barriers to full unification are identified: discrepancies in approaches to identification, impurity control, standardization of active ingredient content, and the issue of interchangeability of pharmacopoeial standard samples. The author concludes that a uniform global quality standard remains a long-term goal, while regional harmonization within the EAEU represents an effective and practical step toward converging regulatory requirements.

INTRODUCTION. The State Pharmacopoeia of the Russian Federation (SP RF) is the key set of mandatory requirements for the drug quality. Its development vectors are not limited to improving the legal framework but also take into account the key trends of the regional pharmacopoeia — the Pharmacopoeia of the Eurasian Economic Union (Ph. EAEU). Regular analysis of the current state of the pharmacopoeia and identification of the most promising and relevant growth vectors is a prerequisite for ensuring sustainable and consistent improvement of pharmacopoeial approaches and requirements.
AIM. This study aimed to identify potential growth vectors of the SP RF, edition XV, given the development of a common pharmaceutical market within the EAEU.
DISCUSSION. We analyzed key changes in the legal framework that took place at the national and regional (EAEU) levels, including the updated Procedure for developing SP RF monographs that explicitly considers Ph. EAEU requirements. The study also examined the Development strategy for EAEU Pharmacopoeia that outlines the basic principles, the Strategy objective, the stages of its implementation, and the necessary measures. The document includes relevant data on the provisions of SP RF, edition XV, and offers promising growth vectors for compendial requirements.
CONCLUSIONS. Considering the main trends in the development of the EAEU Pharmacopoeia, the following growth vectors for pharmacopoeial quality standards can be identified: updating the nomenclature and content of general pharmacopoeial monographs for medicinal products and their dosage forms; extending harmonization to align approaches with the pharmacopoeias of states friendly to the Russian Federation; implementing instrumental, accurate, and selective methods and techniques, in order to replace animal testing as well.
INTRODUCTION. Loperamide hydrochloride medicinal products are registered in the Russian Federation in the dosage forms of “tablets” and “capsules”. Since the State Pharmacopoeia of the Russian Federation (SP RF) does not include pharmacopoeial articles on dosage forms of loperamide hydrochloride, it seems relevant to conduct a comparative analysis and summarize the requirements of foreign pharmacopoeias for quality control of medicinal products based on loperamide hydrochloride.
AIM. Analysis and generalization of quality requirements for loperamide hydrochloride medicinal products for the preparation of recommendations for the compilation of specifications for drugs containing loperamide hydrochloride in tablet and capsule dosage forms, and recommendations for updating the current pharmacopoeial article for the pharmaceutical substance loperamide hydrochloride.
MATERIALS AND METHODS. A study of the quality requirements to pharmaceutical substances of loperamide hydrochloride in the dosage forms of tablets and capsules has been conducted using SP RF, the European Pharmacopoeia (Ph. Eur.), the British Pharmacopoeia (BP), the United States Pharmacopoeia (USP), the Indian Pharmacopoeia (IP), the Pharmacopoeia of the People’s Republic of China (ChP), the Korean Pharmacopoeia (KP), and the International Pharmacopoeia (Ph. Int.). The materials of registration dossiers for loperamide hydrochloride have been reviewed.
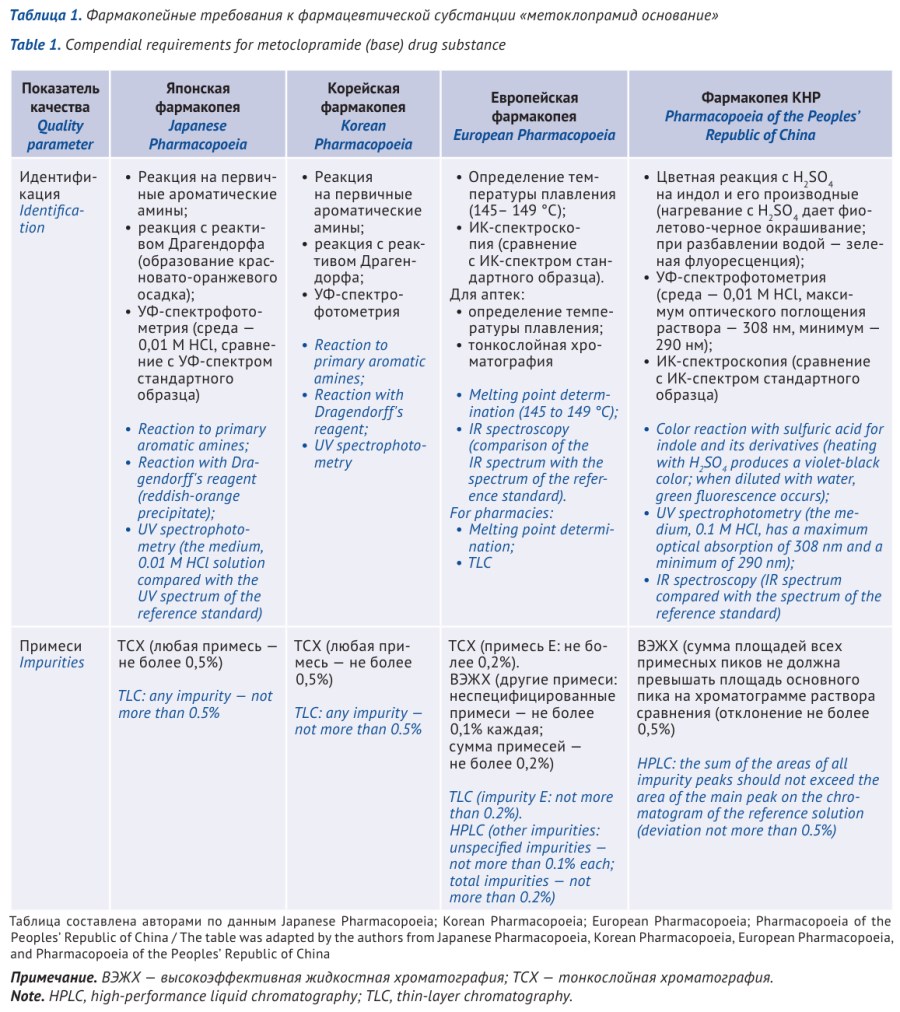
RESULTS. Loperamide hydrochloride is not described in the SP RF. The pharmacopoeial monograph for the pharmaceutical substance loperamide hydrochloride is included in the SP RF (15th ed.), as well as in Ph. Eur., BP, USP, IP, ChP, KP, and Ph. Int. A comparative analysis of pharmacopoeial quality standards for the pharmaceutical substance loperamide hydrochloride revealed that FS.2.1.0613 “Loperamide hydrochloride” of the SP RF is largely harmonized with Ph. Eur., its requirements are among the most stringent and employ more sophisticated methods for identifying and controlling impurities compared to USP, IP, ChP, and KP. However, when revising the pharmacopoeial articles, the combination of methods for identifying loperamide should be optimized, and the control of heavy metals by a semi-quantitative method should be excluded, replacing it with an assessment of the risks of elemental impurity content.
CONCLUSIONS. In preparing specifications for loperamide tablets and capsules, recommendations were provided for the use of combinations of identification methods, potential differences in dissolution testing conditions and standards were identified, and specified impurities were defined. Using various modifications of loperamide as an example, areas for improving the standardization of tablets are demonstrated, including the need for both disintegration and dissolution testing. The feasibility of monitoring the dosage uniformity of lyophilized tablets, regardless of dosage, using the calculated mass method is substantiated.
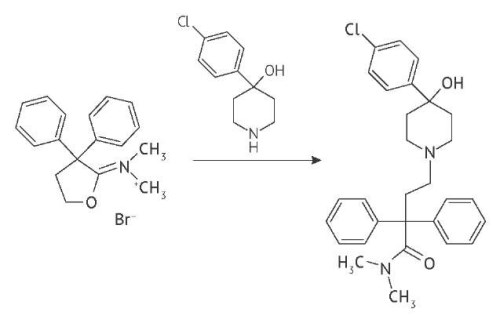
INTRODUCTION. Metoclopramide is a medicinal product widely used as an antiemetic. The pharmaceutical industry applies metoclopramide in the form of base or hydrochloride monohydrate. The pharmacopoeial monograph for metoclopramide hydrochloride monohydrate drug substance was included in the State Pharmacopoeia of the Russian Federation (SP RF); however, SP RF specifies no quality requirements for metoclopramide-based medicinal products. In order to develop a relevant monograph, it seems necessary to systematize the current foreign and Russian compendial requirements.
AIM. This study aimed to develop an approach to quality control of metoclopramide drug substance (hydrochloride and base) and a medicinal product in the dosage form of tablets.
MATERIALS AND METHODS. The study used comparative data analysis and content
analysis. Compendial requirements of SP RF, Edition XV, and the leading foreign pharmacopoeias — European Pharmacopoeia (Ph. Eur.), British Pharmacopoeia (BP), United States Pharmacopeia (USP), Indian Pharmacopoeia (IP), Pharmacopoeia of the People's Republic of China (ChP), Japanese Pharmacopoeia (JP), and Korean Pharmacopoeia (KR) were analyzed.
RESULTS. We substantiated the choice of quality parameters, analytical methods and acceptability criteria used to draft the guidelines on preparing specifications for metoclopramide hydrochloride and base, as well as tablets. The requirements found in SP RF and foreign pharmacopeias were compared, as well as sections of registration dossier for drug substance and tablets covering key quality indicators: Identification, Impurities, and Assay for the substance and tablets; Dissolution (additionally for tablets). Recommendations were given to include quality parameters and analytical methods for the monograph on metoclopramide base. It was proposed to reduce the number of identification methods included in the monograph on Metoclopramide hydrochloride monohydrate.
CONCLUSIONS. Сomparative analysis of compendial requirements has made it possible to offer a unified approach to the formation of quality requirements for metoclopramide products (drug substances and tablets). The approach of the British Pharmacopoeia is recommended for rationing any single impurity and total impurities. It is also recommended to include Metoclopramide base monograph in the SP RF.
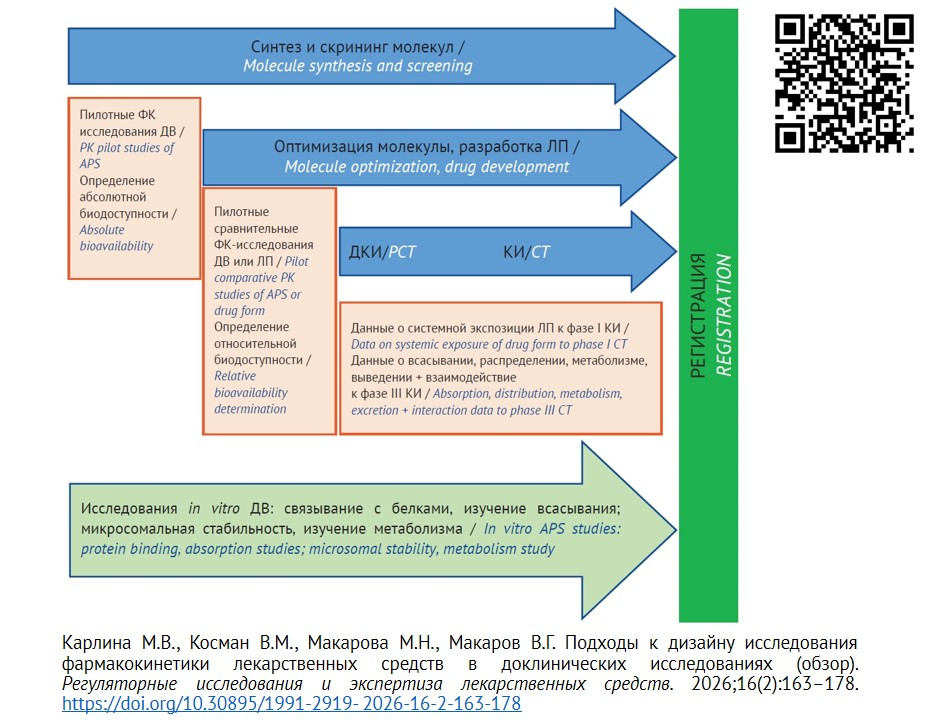
INTRODUCTION. According to drug registration requirements, a registration dossier should include the data on preclinical trials of drug pharmacokinetics. However, the scope of data provided for various drugs groups, as well as pharmacokinetics elements to be studied at each stage of the product life cycle is still a relevant issue.
AIM. This study aimed to analyze literature data, Russian and foreign guidelines on preclinical trials of drug pharmacokinetics to choose an optimal strategy of data collection at various stages of drug life cycle.
DISCUSSION. The research materials included regulatory documents, guidelines on preclinical trials, scientific articles and other publicly available sources (including electronic RSCI databases (eLIBRARY.RU), PubMed, and Web of Science). We analyzed the main approaches to planning pharmacokinetic trials for various drugs (original, generic, biological drugs, etc.), including the type and number of laboratory animals, the doses studied and the time points of biomaterial sampling.
CONCLUSIONS. Preclinical pharmacokinetic trials are warranted for optimization of active pharmaceutical substance molecules, selection of the dosage form to study various drug groups, predicting pharmacokinetic parameters in humans, reducing time costs and risks when developing safe and effective drugs. Designs of pharmacokinetic trials were proposed at the stage of molecule screening, molecule optimization and selection of the drug forms for the research of various drug groups.
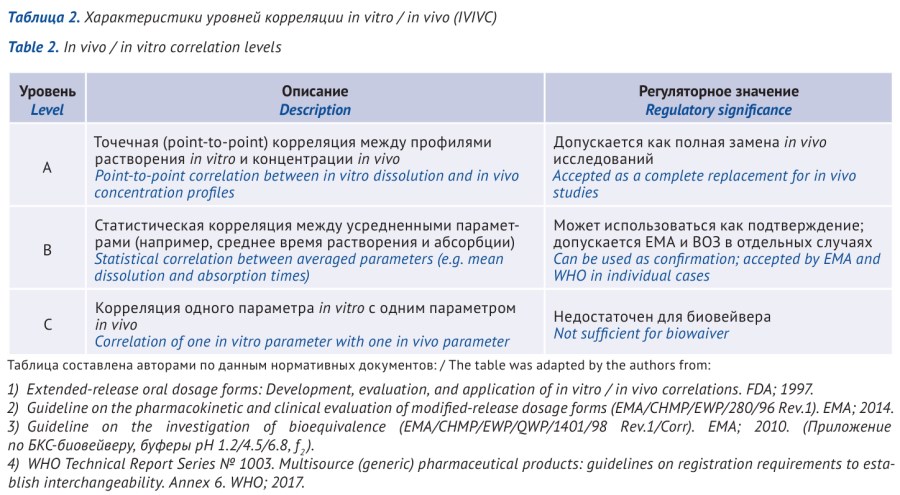
INRODUCTION. Biowaiver procedure based on the biopharmaceutics classification system (BCS) is widely used in regulatory practice when assessing the bioequivalence of generic medicinal products (GMPs). The use of the biowaiver is aimed at reducing the rate of in vivo studies while maintaining the requirements for GMP quality, safety, and efficacy. Over the recent years, additional analytical and modeling tools used in bioequivalence assessment have been actively discussed in the scientific literature. This requires a clear distinction between regulatory requirements and research approaches, including in vitro / in vivo correlations (IVIVC), physiologically based pharmacokinetic (PBPK) modeling, and artificial intelligence (AI) algorithms.
AIM. This study aimed to compare the existing regulatory approaches to the application of the BCS-based biowaiver procedure, with an emphasis on current requirements used to assess GMP bioequivalence.
DISCUSSION. In all considered regulatory systems, the use of the biowaiver procedure is limited to active pharmaceutical ingredients (APIs), BCS classes I and III, with strict adherence to solubility criteria and comparability of dissolution profiles. The division of Class II into subclasses IIa and IIb is not stipulated in any official guidelines and is used exclusively in the scientific literature for the research purposes. It has been established that IVIVC, PBPK models, and AI technologies are considered by regulatory authorities as supporting scientific tools and do not have independent status as an evidence base used to renounce the use of in vivo studies.
CONCLUSIONS. The findings confirm the lack of fundamental regulatory differences between approaches to the biowaiver application. Proposals for expanding biowaiver practice identified in the references relate to scientific controversy without reflecting current regulatory practice. The work forms a methodologically correct basis for the interpretation of regulatory requirements and further research in the area of biopharmaceutical GMPs evaluation.
INTRODUCTION. Vancomycin is a tricyclic glycopeptide antibiotic produced by Amycolatopsis orientalis. To determine the activity of antibiotics obtained by biosynthesis, in particular vancomycin, chemical or physicochemical identification of the active compound is not available; instead, two pharmacopeial methods are used: the agar diffusion and the turbidimetric method. Both methods are based on inhibiting the growth of the test microorganism under the appropriate experimental conditions. Turbidimetric method has several advantages over the agar diffusion method for parenteral preparations used in the form of solutions, as it is more sensitive to low concentrations, more cost-effective and takes less time to obtain the results. The State Pharmacopoeia of the Russian Federation (SP RF) provides one method for determining the activity of antibiotics — the agar diffusion method. Previously, turbidimetric method was developed to determine vancomycin activity.
AIM. This study aimed to evaluate the determination results of vancomycin activity obtained using turbidimetric method in order to use it as an alternative to the agar diffusion method.
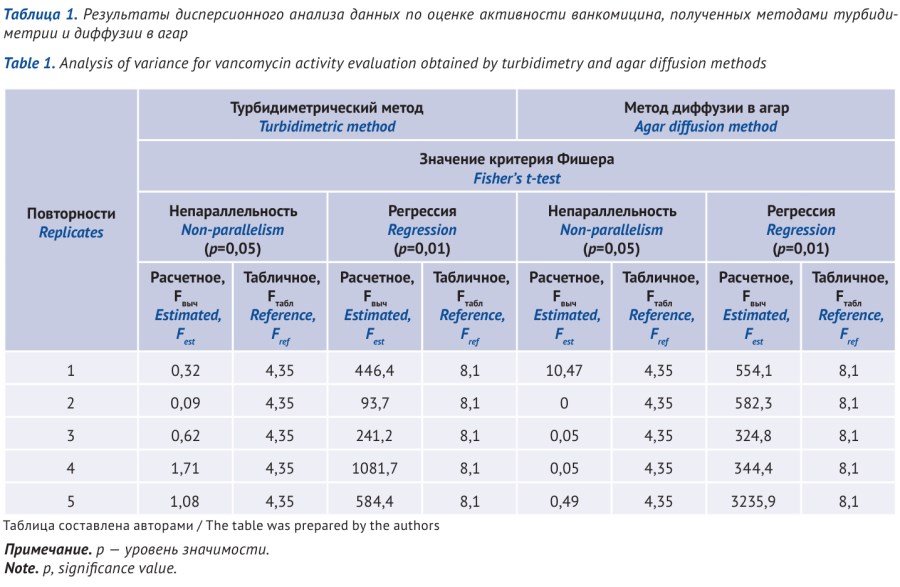
MATERIALS AND METHODS. The test conditions for the method using agar diffusion corresponded to SP RF: test-microorganism, seed dose: Bacillus subtilis NCTC 10400/ ATCC 6633 (Becton Dickinson France S.A.S.), 20×107 spores per ml medium; purified sterile water for the preparation of basic solutions, sterile buffer No. 4 for process solutions. The turbidimetric method included Staphylococcus aureus NCTC 7447/ ATCC 6538P (Becton Dickinson France S.A.S), purified sterile water, and solvent for process solutions — sterile buffer solution, pH 8.0 (adapted from the European Pharmacopoeia). A culture medium developed at the Scientific Centre for Expert Evaluation of Medicinal Products was used to determine the activity of aminoglycosides. The activity was determined by comparing with a reference standard of vancomycin hydrochloride EP CRS banth 4 for microbiological analyses. For each test (agar diffusion and turbidimetry), five accurately weighed samples were taken with an accuracy of 0.0001 g of the test sample; each sample was analyzed in three replicates.
RESULTS. The calculated main parameters of the analysis of variance, Non-parallelism and Regression of the results of the test and reference solutions, confirmed the adequacy of the data obtained using both methods. Comparison of the sample variances of the two methods (turbidimetry and agar diffusion) according to Fisher’s test and Bartlett’s test, the calculated value χ2 (P=95%, f=1), and comparison of the mean values according to the Student’s t-test confirmed that both samples belonged to the same general sample. The systematic error and uncertainty for the turbidimetric method was significantly higher than the agar diffusion: ε 1.5% and ε 0.77%, δ=4.7% and δ=0.97%, respectively.
CONCLUSIONS. The turbidimetric method can be deemed as an alternative for determining vancomycin activity. However, in order to minimize the influence of the human factor and the external study conditions, a preliminary standardization of conditions, with a subsequent implementation control is warranted for the turbidimetric method.
INTRODUCTION. Caraway fruits are used in the treatment of irritable bowel syndrome and dyspeptic disorders due to their pronounced antispasmodic, antimicrobial, and carminative effects. The antispasmodic activity of caraway seeds isprimarily due to their essential oil content, with carvone being the predominant component. The European Pharmacopoeia 11th edition regulates the carvone content in essential oils, while the State Pharmacopoeia of the Russian Federation XIV edition does not standardize caraway fruits based on their carvone content.
AIM. To develop and validate a spectrophotometric method for the quantification of carvone in the essential oil of caraway fruits and to verify the results of the spectrophotometric determination using gas chromatography — mass spectrometry.
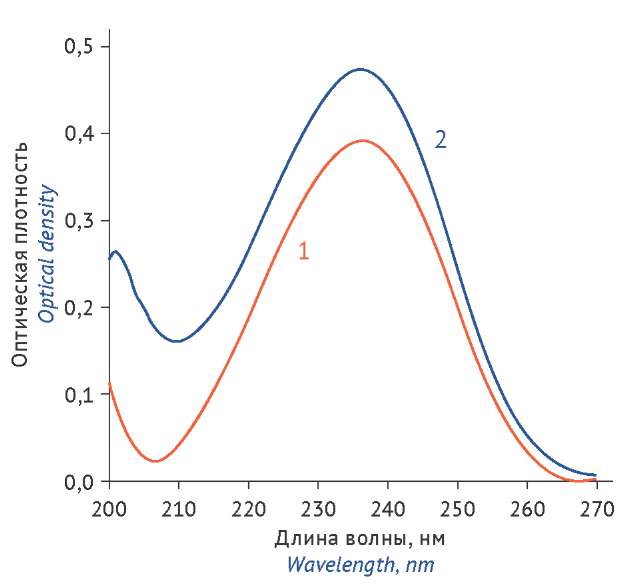
MATERIALS AND METHODS. The study utilized caraway fruits (Carum carvi L.) manufactured by Firma Zdorovie LLC (batch 031224; expiry November, 2027), a carvone reference standard with a purity of 98.5% (Fluka), a UNICO 2802 spectrophotometer (United Products & Instruments, Inc.), ADV-200M analytical balance (GOSMETR), and a GC-MS QP2010 Ultra gas chromatograph (Shimadzu) coupled with a mass-selective detector. For spectrophotometric analysis, aliquots of the essential oil and the carvone reference standard were dissolved in ethanol, 95% (v/v). The absorbance of the solutions was measured at a wavelength of 236 nm in 10 mm path length cells. The results obtained by the spectrophotometric method were confirmed using gas chromatography.
RESULTS. Validation of the spectrophotometric method for the quantitative
determination of carvone, carried out in accordance with the requirements of
OFS.1.1.0012 “Validation of analytical methods” of the State Pharmacopoeia of
the Russian Federation, XV ed., showed its specificity, linearity, precision, and accuracy. The results are not affected by systematic error. The relative standard deviation (RSD) value does not exceed 2.0%. The results of the spectrophotometric determination of carvone (75.7±0.9%) correlate with the results of the gas-liquid chromatography analysis (75.2±0.8%).
CONCLUSIONS. The developed analytical procedure for the quantitative determination of carvone can be employed for the standardization of caraway fruits by introducing an additional quality parameter for their essential oil in the “Assay” section. It is also suitable for the standardization of caraway essential oil when used as a substance for the manufacture of medicinal products.
INTRODUCTION. The content of pesticides in pharmaceutical raw materials is regulated by the Russian legislation, namely General pharmacopoeial monograph, OFS.1.5.3.0011 Determination of Residual Pesticides in Pharmaceutical Raw Materials and Medicinal Plant Preparations of the State Pharmacopoeia of the Russian Federation, XV edition (SP RF). Residual pesticides are one of safety parameters for medicinal plant raw materials. SP RF stipulates the content of 65 compounds; however, the pesticide market is actively expanding (at least 10% per year), while not all compounds used are subject to quality control. Pesticides widely used in agriculture to control weeds, such as dicamba (3,6-dichloro-2-methoxybenzoic acid), a derivative of chlorobenzoic acid, are of particular interest. Dicamba is used to produce Lintur and Dialen Super, formulations for weed eradication.
AIM. This study aimed to develop a selective identification method for dicamba herbicide in the pharmaceutical raw materials of Echinacea purpurea for its subsequent introduction into pharmacopoeial standards.
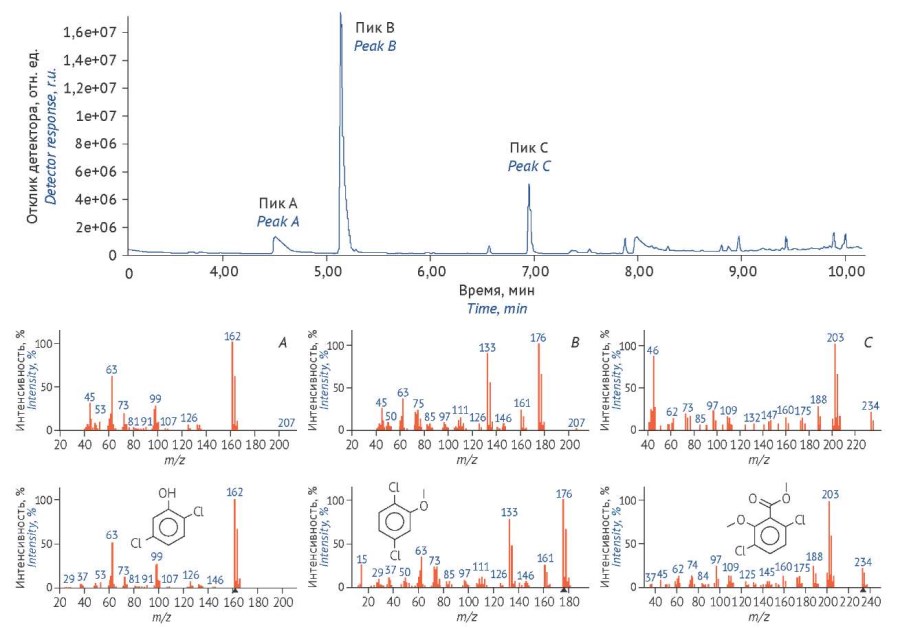
MATERIALS AND METHODS. The study used a reference standard of dicamba (Kit-54, pesticide 4-4600, Polycience Corporation); the pharmaceutical raw material Echinacea purpurea, Livadia strain, cultivated from the seeds (seed manufacturer Aelita, Russia, best before 06/2028); and herbicide mixture Lintur, active substance dicamba 659 g/kg and triasulfuron 41 g/kg, (Gardener’s Green Pharmacy Co. Ltd.). High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) was performed on Agilent Technologies 1260 Infinity modular liquid chromatograph with Agilent Technologies 6420 Triple Quad LS/MS tandem mass spectrometer; gas chromatography-mass spectrometry (GC-MS) was performed on a gas chromatograph connected to QP-2020 monoquadrupole mass spectrometer (Shimadzu). Chromatograms and mass spectra obtained using HPLC-MS/MS were analyzed in Agilent MassHunter Quantitative Analysis B.06.00 databases, GC-MS chromatograms — in Аgilent G1710 MSD Data Analysis ChemStation. NIST MS Search Program 17 Mass Spectral Library was used to analyze mass spectra obtained by GC-MS.
RESULTS. We developed an assay method of dicamba herbicide in pharmaceutical raw materials using HPLC-MS/MS and evaluated the validation parameters (chromatographic system suitability, specificity, linearity, accuracy, and precision). The possibility of using GC-MS method with/without silylation was examined. The maximum permissible value of dicamba herbicide in pharmaceutical raw materials was calculated, and it was concluded that Echinacea purpurea raw materials cultivated using this herbicide do not meet the compendial requirements.
CONCLUSIONS. The findings demonstrate suitability of the HPLC-MS/MS method and the inadequacy of the GC-MS method, both with and without derivatization, for dicamba quantitation in the pharmaceutical raw materials. The developed procedure can be offered as the main method used to quantify dicamba in the pharmaceutical raw materials to be included in the compendial requirements.
INTRODUCTION. Medicines, based on recombinant DNA technology, are widely used in the treatment of cancer, immune-inflammatory, and infectious diseases. Research on the development of new protein-derived drugs, including monoclonal antibodies, as well as methods for quality control of such drugs, is ever increasing. Peptide mapping allows it to verify the primary protein structure, genetic stability, and identify structural changes.
AIM. Systematization of current methodological approaches to the development of peptide mapping methods.
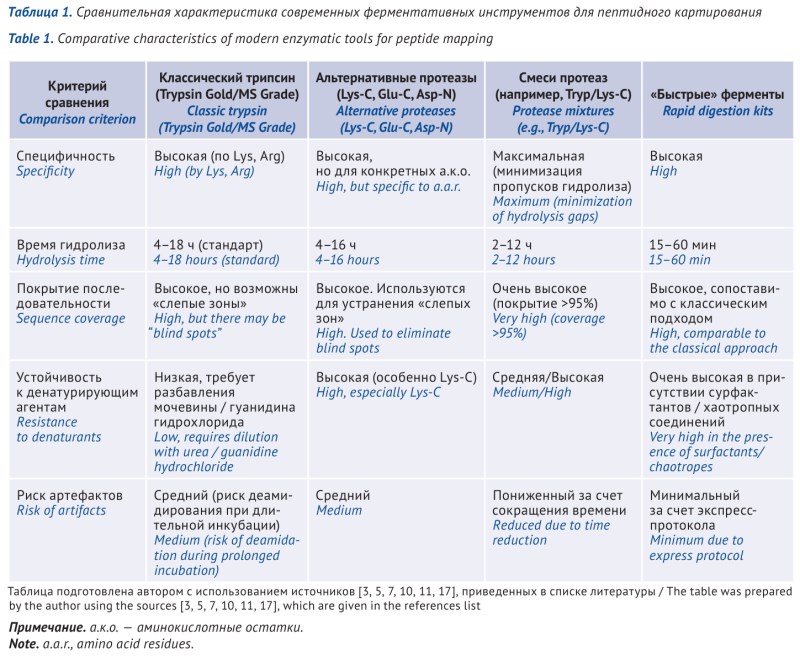
DISCUSSION. One of the main methods for confirming protein authenticity is peptide mapping based on enzymatic hydrolysis of protein to produce a unique set of peptide fragments. Despite the uniqueness of each method, they all share common principles of sample preparation and analysis, as well as regulatory requirements. Method development is a complex, multi-step process. Currently, various enzymes are used for protein cleavage, but trypsin remains the “gold standard”. Ready-made solutions for protein cleavage reactions are becoming increasingly common such as highly specific and reproducible kits, sample preparation approaches based on immobilized enzymes on magnetic particles, and automated procedures. These options minimize errors in manual sample preparation, improve the reproducibility of results, and reduce analysis time.
CONCLUSIONS. Current peptide mapping methodology is evolving towards increased reproducibility and efficiency through the introduction of standardized and automated sample preparation solutions while maintaining trypsin as the primary enzyme for protein cleavage. Information on approaches used will enable researchers to navigate the diversity of available analytical solutions, accelerate method development, and ensure reliable quality control of protein-based medicines.
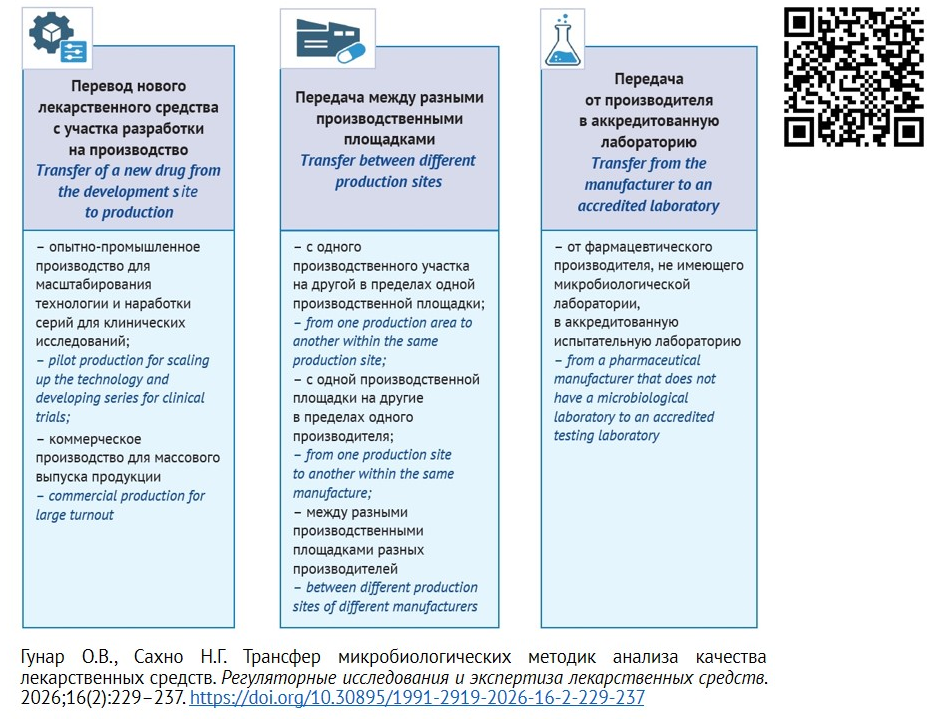
INTRODUCTION. The transfer of microbiological methods is an important aspect of analytical methods transfer as part of technology transfer between production sites of the same or different pharmaceutical companies, as well as to contract laboratories. The regulatory and organizational aspects of analytical method transfer are clearly defined and described in the scientific literature, while the transfer of microbiological methods, when not specifically identified as a separate area, often presents a number of challenges, for instance lack of specialized premises and qualified staff, permission to work with pathogenic microorganisms, etc.
AIM. Developing an algorithm for the transfer of microbiological methods for drug quality analysis, taking into account the specifics of individual transfer procedure steps.
DISCUSSION. Method transfer is a mutually beneficial process for both the receiving and the transferring parties. When preparing for the transfer of a microbiological method, the parties analyze and assess quality risks across a number of criteria (lab equipment, staff, QMS documentation, etc.), consider the possibility of combining technologies, analyze discrepancies, and make any necessary changes. A transfer plan is then developed; it includes methodological specifications for validation testing, parameters, and acceptance criteria. The microbiological method transfer algorithm requires certified facilities, a license to work with microorganisms of pathogenicity groups 3–4, and trained staff specialized in microbiology and microbial biosafety. The transferring and receiving parties can jointly implement staff training programs. Official pharmacopoeial methods do not require transfer. Using alternative microbiological methods extensive comparative validation studies are required.
CONCLUSIONS. The transfer of microbiological methods is the most critical part of the transfer process at a pharmaceutical company, which is currently based on OFS.1.1.0030 GF RF, OFS 2.3.16.0 FEAEU, and EEC Guideline No. 11 dated June 8, 2021. Reasonable amendments to the transfer procedure may be made regarding the functioning of a specific laboratory, including microbiological monitoring, cleaning validation, etc.

ISSN 3034-3453 (Online)





























