


PRECLINICAL TRIALS: A COURSE TOWARDS TRANSLATIONALITY

The concept of translational gap implies that the intensive development of basic biomedical research at the turn of the 20th and 21st centuries has not, until recently, been accompanied by a proportional increase in the number of innovative drugs, as well as diagnostic and therapeutic technologies that have found application in clinical practice. Mikhail M. Galagudza, Director of the Institute of Experimental Medicine at the Almazov National Medical Research Center of the Russian Ministry of Healthcare, Doctor of Medical Sciences, Professor and Corresponding Member of the Russian Academy of Sciences, shares his thoughts on the causes of the translational crisis and ways of its resolution.
INTRODUCTION. Currently, the Eurasian Economic Union (EAEU) lacks guidelines for documenting preclinical studies. At the same time, proper registration of raw data is necessary to confirm the quality of the preclinical results obtained.
AIM. This study aimed at introducing a documentation procedure for preclinical studies that would cover documents from the initial study application to the final report and provide for preclinical data transfer to the marketing authorisation dossier.
MATERIALS AND METHODS. The authors opted for information analysis as the method of research. All recommendations for documenting preclinical studies were formulated in accordance with the EAEU Good Laboratory Practice (GLP) requirements applicable to the medicinal product lifecycle and the work of preclinical study sites using experimental animals.
RESULTS. The general EAEU recommendations for the conduct of animal studies are not sufficient to achieve adequate quality of preclinical studies. This article proposes a procedure for complete documentation of preclinical studies, with all documents following the documentation requirements of the EAEU GLP for each study stage. When developing the necessary forms and documents, each study site should operate under its own quality management system and consider the preclinical study specifics on a case-by-case basis. The preparation of any given document should ensure compliance with the GLP principles and guarantee the completeness and integrity of the data obtained.
CONCLUSIONS. Study sites can implement the proposed documentation procedure to design and conduct preclinical studies in accordance with the regulatory requirements that determine the role and responsibilities of the study director and the conduct of inspections by the quality assurance unit.
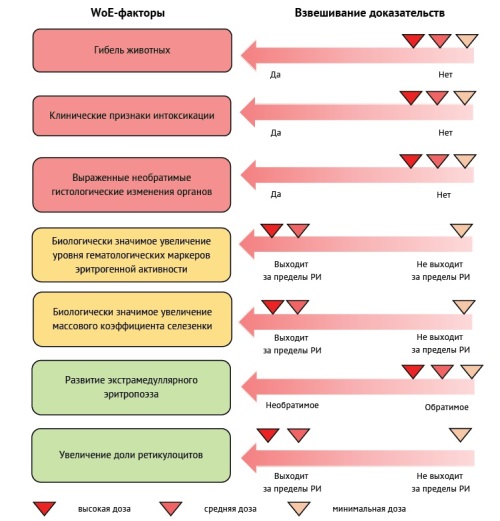
INTRODUCTION. Toxicological studies of new pharmaceuticals in laboratory animals represent an obligatory stage in drug risk assessment, designed to identify toxic effects, their potential reversibility, and dependence on dose and/or systemic exposure. The primary quantitative outcome of these studies is the no-observed-adverse-effect level (NOAEL), whose key practical application involves determining the starting dose for early-phase human clinical trials. The very definition of NOAEL raises the fundamental question: which experimental changes should be classified as adverse? Toxicology study designs are complex, incorporating a comprehensive range of evaluation parameters including physiological, instrumental, clinical laboratory, and pathological morphological assessments. Upon study completion, researchers accumulate extensive datasets requiring rigorous scientific analysis and evidence-based interpretation of all detected changes.
AIM. The aim of this study is to develop a comprehensive methodology for interpreting toxicological data to enhance objectivity in NOAEL determination.
DISCUSSION. Due to the lack of terminological rigor in describing the NOAEL dose, the present work attempts to unify this term. Particular attention is given to the issue of biological significance of observed changes, as well as the description of criteria for determining their adversity. The developed scheme for interpreting experimental data encompasses three consecutive stages: analysis of the relationship between the observed effect and the administration of the test object; assessment of the effect size and/or degree of changes; determination of the nature of the identified changes in terms of their adversity, including evaluation of their reversibility, pharmacodynamic acceptability, potential for developing an adaptive response, and others. The cornerstone of the proposed methodology is the Weight-of-Evidence principle, which enables ranking of identified adverse effects according to their significance and their integration into a unified assessment system.
CONCLUSIONS. The developed integrated approach, based on multifactorial data analysis (including quantitative and qualitative indicators) and the Weight-of-Evidence assessment principle, can enhance the objectivity of evaluating observed effects and determining the No Observed Adverse Effect Level (NOAEL).
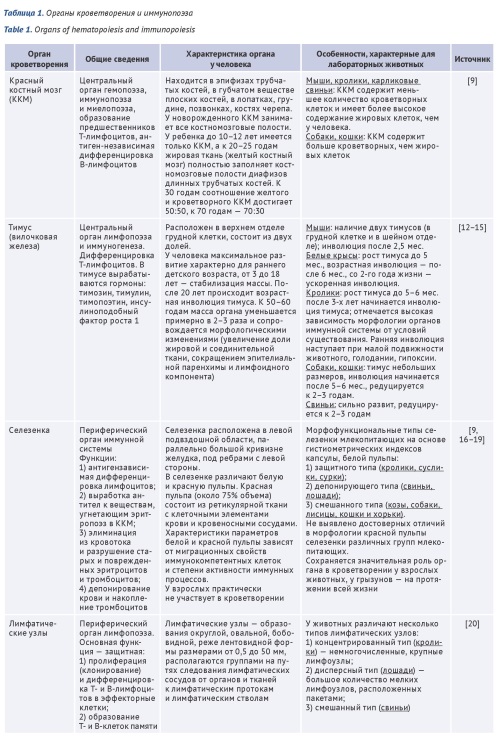
INTRODUCTION. The blood contains a large quantity of active cells of the organism and is a basic target for potentially toxic substances. The regulatory documents of the Russian Federation and international recommendations provide a list of mandatory biochemical and hematological parameters, but they are insufficient for predicting drug-induced hepatotoxicity in vivo during preclinical studies. A review of new data on this issue and an update of the list of biomarkers will expand the capabilities for monitoring the condition of laboratory animals, enhance the sensitivity and specificity of assessing toxic effects on hematopoiesis in preclinical studies, and thereby contribute to improving the safety of medicinal products and the effectiveness of therapy. The review consists of two parts.
AIM. The study aimed to identify the differences between the hematopoietic organs of humans and laboratory animals in order to develop recommendations for preclinical studies using animal blood as a biomaterial.
DISCUSSION. The first part of the review shows the characteristics of the hematopoietic organs of humans and laboratory animals, indicating their characteristics compared to humans. It is discussed of mechanisms of hemotoxicity of various medicinal products. The paper discusses the mechanisms of development of hematotoxicity of drugs, among which 4 main ones can be distinguished: cytotoxic effects on hematopoietic progenitor cells; direct damage to mature cells; indirect damage to blood or bone marrow cells due to undesirable immune reactions against and changes in the activity of cell surface receptors; change in the activity of enzyme systems necessary for the normal functioning of cells. Data on clinical blood analysis parameters of 8 species of laboratory animals: rodents (mouse, rat, hamster, guinea pig) and non-rodents (rabbit, macaque, pygmy pig, ferret) are summarized.
CONCLUSIONS. Some features in the structure and function of the hematopoietic organs compared to humans can lead to significant differences in the toxicological profile of the drug. It should be noted that a clinical blood test allows us to assess a significant number of manifestations of hematotoxicity of drugs that directly affect blood cells and their precursors. With an indirect effect of the medicinal product (through enzyme systems, effect on progenitor cells, appearance of antibodies, etc.), these data are insufficient and the use of additional markers is necessary in order to increase the predictive value of preclinical studies and the comparability of data with clinical studies.
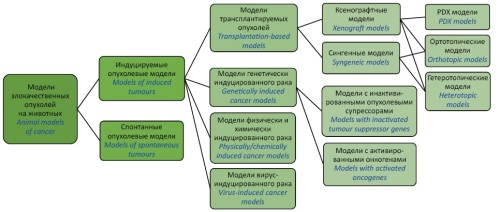
INTRODUCTION. The main risk to the clinical translatability of preclinical results for anticancer medicinal products is posed by the difficulty of simulating clinical conditions in an experimental model. With only 5% of product candidates proving clinically effective, the search for new approaches to the preclinical development of anticancer medicinal products is currently an active area of research in medicine.
AIM. This study aimed to provide methodological support for planning experiments with modelling of neoplastic processes through analysis and classification of the methods used in preclinical studies of the efficacy of anticancer medicinal products in vivo.
DISCUSSION. This article reviews the development of animal tumour models and the selection of cell lines and their testing for tumourigenicity and viability on a step-by-step basis. According to the study results, imaging systems, vital staining, and fluorescence- and luminescence-based methods can be used to assess the efficacy of anticancer medicinal products in both solid tumour models and haematological malignancy models. The article presents a schematic representation of the main types of mouse cancer models. However, no single animal species is universally suitable for in vivo cancer modelling. Researchers selecting models and considering their advantages and disadvantages should pay special attention to the similarity of disease mechanisms in animal models and humans at the tissue and molecular level, keeping in mind the aims of their research.
CONCLUSIONS. The results of this comparative analysis of methods for preclinical efficacy evaluation of anticancer medicinal products are essential for designing experimental studies and ensuring the reliability of the results obtained. Choosing the correct research method will increase the chances of obtaining experimental data that can be successfully translated into clinical practice.
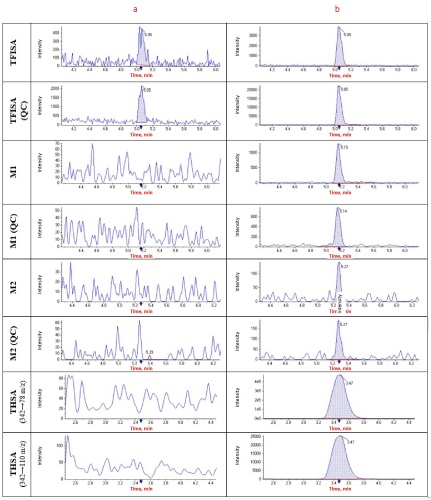
INTRODUCTION. This article continues a series of publications on the pharmacokinetics of 5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (TFISA), a novel compound for the treatment of open-angle glaucoma. The distribution of TFISA and its metabolites in rat organs and tissues has not been previously studied in preclinical trials.
AIM. This study aimed to assess the tissue distribution and bioavailability of 5-[5-(trifluoromethyl)-1,2-oxazol-3-yl]-furan-2-sulfonamide and its metabolites in rat organs and tissues and to validate the analytical procedures developed for this purpose. MATERIALS AND METHODS. The study used 60 male Wistar rats. TFISA was administered by bilateral ocular instillation of 1% ophthalmic suspension at a dose of 40 μL (approximately 3.7 mg/kg). Tissue samples (liver, kidney, lung, brain, heart, spleen, skin, muscle tissue, and eyes) were collected 1, 2, 4, 8, 12, 24, 48, 72, 144, and 216 h after instillation (from 6 rats at each time point). The samples were immediately homogenised using methanol and were stabilised with ascorbic acid solutions. High-performance liquid chromatography with tandem mass spectrometric detection was used to quantify TFISA, N-hydroxy-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]furan-2-sulfonamide (M1), and N-acetyl-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (M2) in homogenised organ and tissue samples.
RESULTS. This study involved full validation of the analytical procedures developed for the quantitative determination of TFISA and its metabolites, which was conducted separately for eye tissues and other biological samples. The tissue bioavailability (ft) of TFISA decreased from 13.0 to 0.7 in the following order: eye tissues (administration and action site) > spleen > lungs ≥ heart ≥ liver > kidneys > brain > skin ≥ muscles. The ft values for M1 decreased from 52.0 to 2.5 in the following order: spleen ≥ lungs ≥ heart ≥ liver > kidneys > brain > muscles ≥ eye tissues > skin. The ft values for M2 were lower than those for TFISA and M1 and decreased from 6.4 to 0.3 in the following order: liver ≥ kidneys > heart > lungs > eye tissues > skin > spleen ≥ muscles > brain.
CONCLUSION. The validated bioassays have been successfully applied to study the distribution of TFISA and its metabolites in male rats. TFISA best penetrates into the eyes and well-vascularised organs. M1 is highly bioavailable in the spleen, heart, and lungs. M2 shows the highest bioavailability in the liver and kidneys of rats.
HERBAL MEDICINAL PRODUCTS
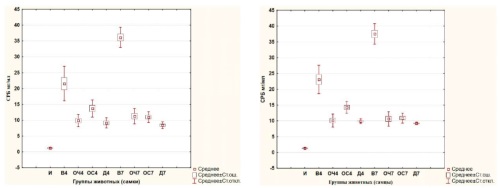
INTRODUCTION. Flavonoids contained in many plant species inhibit the induction of cytokines and arachidonic acid metabolites, which are tissue mediators of inflammation, thus exhibiting an anti-inflammatory effect. The leaves of black and gray alder contain flavonoids, phenolic carboxylic acids, tannins and can be considered as a new type of herbal medicinal raw materials.
AIM. Evaluation of the anti-inflammatory effect of decoctions and gels containing the sum of biologically active substances of black alder (Alnus glutinosa (L.) Gaertn.) and gray alder (Alnus incana (L.) Moench.) leaves on laboratory animals using various models of inflammation.
MATERIALS AND METHODS. The anti-inflammatory activity of aqueous extracts of black alder and grey alder leaves, as well as gels containing their alcoholic extracts, was studied in models of generalized and local inflammation induced by the administration of a 1% λ-carrageenan solution to Wistar rats (female and male). The anti-inflammatory effect in generalized inflammation was assessed by blood biochemical parameters (C-reactive protein, alanine aminotransferase, aspartate aminotransferase, γ-glutamate transferase), in case of local inflammation — by changes in the volume and mass of rat paws.
RESULTS. On the model of generalized carrageenan inflammation, the effectiveness of aqueous extracts of black and gray alder leaves was evaluated on days 4 and 7. It was found that introducing aqueous extracts determines a statistically significant decrease in the specific inflammation indicator — level of C-reactive protein in animals receiving an infusion of black alder and grey alder leaves of intragastric introduction. Using models of local carrageenan inflammation, it was established that under the influence of infusions and gels containing biologically active substances of black and gray alder leaves, there was a statistically significant decrease in the increase in weight and paw diameters compared to control groups. The edema inhibition index (calculated by weight) for the gel containing black alder leaf tincture based on 60% ethyl alcohol was the highest and amounted to 57.95% in females and 56.53% in males, for the gel containing grey alder leaf tincture based on 70% ethyl alcohol — 56.78% in males, 52.02% in females.
CONCLUSIONS. It has been proven the anti-inflammatory activity of aqueous and alcoholic extracts from black alder and gray alder leaves in models of generalized and local inflammation. Dosage forms (gels) containing alcohol extracts from the leaves of black alder and gray alder also have an anti-inflammatory effect in conditions of induced local inflammation. Also it has been proven the anti-inflammatory effect of biologically active substances of black alder and gray alder on laboratory animals using models of generalized and local inflammation.
DEVELOPMENT AND VALIDATION OF RESEARCH METHODS
INTRODUCTION. Modern methods for determining particle sizes with the necessary reliability make it possible to study the granulometric composition of medicines and to assess the potential risks in the medical use of medicines in the presence of subvisible particles. This is especially necessary in relation to injectable, inhalation, ophthalmic forms of medicines. Each of the known methods has its own characteristics and the scope of its application.
AIM. Systematization of methods for determining the size of subvisible particles in medicines.
DISCUSSION. The paper considers the most commonly used methods for determining the size of subvisible particles and particle size distribution. The paper considers the most commonly used methods for determining the size of subvisible particles and particle size distribution. The laser diffraction method is used to determine the total number of particles in a sample and the particle size distribution of both the total volume of all particles and the total number of all particles. Dynamic light scattering is mainly used to estimate the size of subvisible dispersed particles suspended in solution. Both methods are based on the use of laser light scattered by particles of the test sample, while the principles of operation of the equipment are different. To identify subvisible particles, the presence of which is undesirable in medicines, the counting light blocking method and the microscopy method are provided. The microscopy) method, based on counting subvisible particles on the surface of a dried membrane filter after passing drug solutions through the membrane, is used to identify subvisible particles if the counting light blocking method cannot be used. The microscopic method with flow visualization and the scanning electron microscopy method are not used for routine control of medicines, but are important additional tools for determining the features of the granulometric composition and detecting undesirable contamination by foreign particles in medicines.
CONCLUSION. The choice of method depends on the task and the object of the study. When applying each of the considered methods, the type and size of particles, the technology of the drug, the physico-chemical properties of the test sample, the speed of analysis, the requirements for the accuracy of the method, the availability of equipment and software, and the competence of personnel should be taken into account. In some cases, it is necessary to combine different methods to obtain the most reliable results or to use the most universal methods, in particular, the microscopic method with flow visualization, which will determine the size, shape, and nature of subvisible particles and the content of particles of a specific size in total.
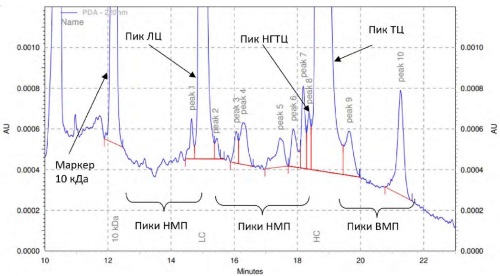
INTRODUCTION. The development of unified techniques for quality control monoclonal antibodies (mAb) is one of the challenges for mAbs quality standardization.
AIM. This study aimed to develop and analyse the possibility of using platform (universal) techniques for estimating the impurity content of high molecular weight compounds and fragments, non-glycosylated molecules in various mAbs by methods of size-exclusion high performance liquid chromatography (SE-HPLC) and capillary electrophoresis methods.
MATERIALS AND METHODS. MAbs-containing medicinal products from 28 different international nonproprietary names of Russian and foreign origin. Studies by SE-HPLC were carried out on Agilent Technologies 1200 series instruments equipped with UV detectors, with data processed via OpenLab software. Capillary gel electrophoresis (CGE) was conducted under both reducing and non-reducing conditions using a PA 800 plus system (Beckman Coulter) with UV diode array detection, and Beckman 32Karat software.
RESULTS. Universal sample preparation protocols and separation conditions were established and validated across 28 mAbs. Retention times and relative migration times of target compounds peaks were determined for each mAb. Comparative analysis demonstrated concordance between platform methods and manufacturers' proprietary methods. Validation of methods for determination of the content of high-molecular weight compounds in mAbs samples by the SE-HPLC method, fragments of mAbs and non-glycosylated variants of heavy chains of mAbs under reducing and non-reducing conditions by the CGE method was performed. The accuracy, precision, and sensitivity of the methods met the established requirements. The validation characteristics obtained for the SE-HPLC method were as follows: precision (RSD) of peak areas was no more than 0.4% for the monomer, up to 8% for aggregate groups, no more than 0.4% for total peak areas, and 0.02% for the relative peak area corresponding to the monomer. The method's linearity was confirmed within the concentration range of 0.5–120%, accuracy within 99.1–102.1%, and the limit of quantification (LOQ) was 0.1%. For the CGE method, the validation characteristics showed precision of no more than 1% for intact immunoglobulin content or heavy and light chain peaks, and no more than 1% for absolute migration times of main peaks. Linearity was confirmed from LOQ to 300% concentration range. The accuracy for both methods ranged from 97.6–103.7%, with LOQ values of 0.5% and 0.75%, respectively.
CONCLUSIONS. The developed methods for assessing the purity are universal for unconjugated mAbs products. Their validation characteristics — including specificity, precision, limit of quantification, analytical range, linearity, and accuracy — meet all established acceptance criteria. These methods can be reliably implemented at any stage of the product lifecycle for this category of medicinal products.

INTRODUCTION. Determination of pyrogenic contaminants in medicines is a critical quality indicator that guarantees the preservation of life and health of patients. In order to control such substances, the biological indicator "Pyrogenicity" was introduced in the middle of the 20th century, which is based on the assessment of the reaction of rabbits to the introduction of the test sample. In accordance with the 3R concept, since 2009, in addition to testing on rabbits, the European Pharmacopoeia has included a monograph of an alternative in vitro method - the Monocyte Activation Test. This test was included in the domestic pharmacopoeia in 2018 (OFS.1.2.4.0016.18 "Monocyte Activation Test"). However, to date, the method has not received wide application among manufacturers either in Russia or abroad due to the peculiarities of the test, related to the need for a significant amount of human blood to obtain monocytes, with a limited choice of reagent kits, etc. The "Pyrogenicity" test is still in demand in the field of determining pyrogenic impurities of any nature (bacterial endotoxins and non-endotoxin pyrogens). For more complete harmonization with European requirements, implementation of the 3R concept related to humane treatment of animals, the introduction of the monocyte activation test is one of the priority tasks.
AIM. Evaluation of the possibility of using the monocyte activation test as a primary test for the determination of non-endotoxin pyrogens.
DISCUSSION. The article describes pharmacopoeial methods for determining pyrogenic substances, analyzes in detail the method for determining pyrogens using human blood cells – monocytes. Currently, the monocyte activation test is the only in vitro test that allows determining the presence of pyrogens of any nature (bacterial endotoxins and non-endotoxin pyrogens). The test is a worthy example of implementing the strategy of replacing animal tests, it allows determining impurities in cases where control using other methods causes difficulties or cannot be performed. The main disadvantage that complicates the implementation of the test in routine control is the limited availability of human blood as a source of monocytes. Possible sources of monocytes that can be used to determine pyrogenicity, including animal blood cells, are discussed. In accordance with the trend to replace the in vivo test – "Pyrogenicity" with an alternative in vitro method, the need to master the application of the method in relation to biological and immunobiological medicinal products is shown.
CONCLUSIONS. Based on the results of the theoretical analysis of the possibility of using the in vitro method as the main method for determining, first of all, non-endotoxin pyrogens, it was established that the world community is seriously determined to refuse the use of animals. The monocyte activation test is included in most pharmacopoeias of the world, as well as in the domestic pharmacopoeia, and is recommended for implementation as the main control.
HERBAL RAW MATERIAL
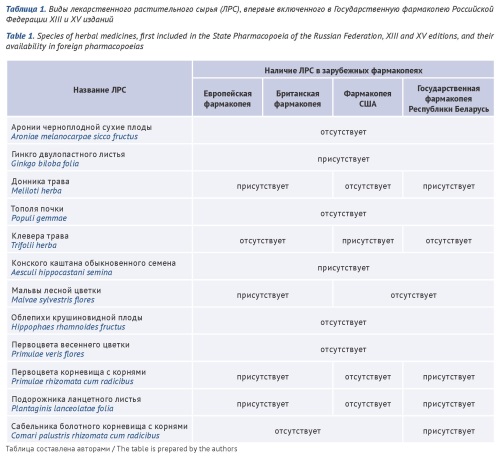
INTRODUCTION. The expansion the nomenclature of herbal drugs and the development of standards for its quality allows us to provide the Russian pharmaceutical industry with new sources of biologically active substances for the creation of effective herbal drug preparations based on them with a favorable safety profile.
AIM. Assessment of changes in the nomenclature and requirements for the quality of herbal drugs in the State Pharmacopoeia of the Russian Federation (Ph. Rus.).
DISCUSSION. Herbal drugs are widely represented in foreign and in our pharmacopoeias. The number of monographs for new species of herbal drugs, as well as updated monographs previously included in the State Pharmacopoeia is growing. If the Ph. USSR X includes 45 monographs for herbal drugs, XI edition – 83 monographs, whereas the Ph. Rus. XV already has 118 monographs. Starting with the Ph. Rus. 13th edition, 106 monographs have been updated and 12 monographs have been included for new species of herbal drugs. It is required a comprehensive analysis of herbal drugs, starting with an assessment of the source of raw materials and ending with proof of the therapeutic efficacy of herbal drug preparations based on it, to include a new quality standard in the Ph. Rus. The requirements for the quality of herbal drugs have undergone changes: in qualitative and in quantitative determination, analysis of substances with therapeutic activity is more and more often used; qualitative reactions have been mainly replaced by chromatographic methods of analysis using standard samples of active substances or markers (active or analytical). For some species of herbal drugs impurities are identified (including unacceptable ones). Requirements with limits tests for the content of toxic substances (heavy metals and arsenic, radionuclides, residual pesticides) have been introduced.
CONCLUSIONS. The inclusion of new species of herbal drugs in Ph. Rus. is accompanied by a significant change in quality requirements and their updating for species of herbal drugs previously included in the pharmacopoeia. Quality control of herbal drugs using new indicators will ensure the authenticity of herbal drugs and increase the safety of their use, which in turn will expand the range of effective and safe herbal drug preparations.

ISSN 3034-3453 (Online)





























