


КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ
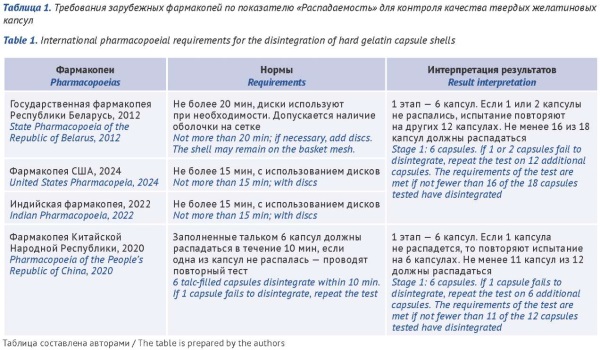
ВВЕДЕНИЕ. Отсутствие национальных требований, регламентирующих качество без содержимого (до их заполнения лекарственным средством), вызывает трудности не только у разработчиков и производителей капсул, но также и у экспертов, осуществляющих оценку представленных на экспертизу материалов. В связи с этим представляется актуальным проведение сравнительного анализа требований к оценке качества ТЖК в России и за рубежом для подготовки проекта фармакопейной статьи на ТЖК без содержимого.
ЦЕЛЬ. Оценка уровня требований к качеству ТЖК, выбор показателей качества, нормативных требований, методов и методик анализа для подготовки проекта фармакопейной статьи на ТЖК.
ОБСУЖДЕНИЕ. По результатам проведенного исследования показано, что растущий интерес производителей к выпуску лекарственных препаратов в форме «капсулы» объясняется преимуществами данной лекарственной формы. В фармакопеи Республики Беларусь (ГФ РБ), Китайской Народной Республики (ФКНР), Индии, Республики Корея и Японии включены монографии на ТЖК без содержимого. Завершено обсуждение соответствующей монографии для Фармакопеи США. Отмечено, что перечень фармакопейных требований к ТЖК включает контроль по показателям «Описание», «Подлинность (желатин, титана диоксид, красители, консерванты)», «Запах», «Средняя масса капсул», «Распадаемость», «Потеря в массе при высушивании», «Микробиологическая чистота», а также контроль посторонних примесей (сульфатной золы, тяжелых металлов и мышьяка) и консервантов (парабенов, серы диоксида). Сравнительный анализ фармакопейных требований показал, что наиболее детализирован подход к контролю качества ТЖК в ГФ РБ и ФКНР. На основе анализа материалов регистрационных досье на 65 препаратов в форме «капсулы», установлено, что в России чаще всего используют ТЖК восьми производителей. Стандарты качества ТЖК этих производителей содержат все общие показатели качества, соответствующие фармакопейным требованиям, а также дополнительные показатели «Мышьяк», «Смазывающие вещества».
ВЫВОДЫ. В результате проведенного сравнительного анализа фармакопейных стандартов качества на ТЖК и стандартов производителей обоснован выбор необходимых показателей качества, норм и требований для включения в проект фармакопейной статьи на ТЖК без содержимого.
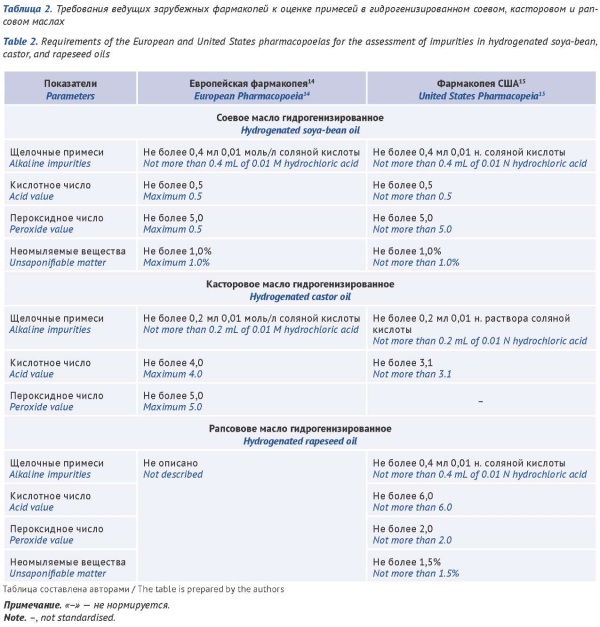
ВВЕДЕНИЕ. В составе лекарственных препаратов (ЛП), представляющих собой масляные растворы, масла жирные растительные (МЖР) могут составлять 50% и более (в некоторых случаях ЛП может на 100% состоять из МЖР). Качество используемого МЖР и протекающие в нем процессы будут оказывать существенное влияние на качество ЛП. Технологический процесс производства, условия хранения и транспортировки ЛП относятся к факторам, влияющим на образование примесей в МЖР, следовательно, актуальным вопросом является рассмотрение национального и международного подходов к оценке примесей в МЖР и ЛП на их основе.
ЦЕЛЬ. Анализ фармакопейных требований к контролю примесей в МЖР и их обобщение в виде рекомендаций для производителей лекарственных препаратов, представляющих собой масляные растворы с содержанием МЖР от 50 до 100%.
ОБСУЖДЕНИЕ. Проведен сравнительный анализ требований Государственной фармакопеи Российской Федерации (ГФ РФ) и ведущих зарубежных фармакопей к контролю примесей в МЖР на примере масел, наиболее часто используемых в качестве растворителей в производстве жидких лекарственных форм (кунжутное, оливковое, подсолнечное, соевое, рапсовое, касторовое). Установлено, что профиль примесей на одноименные МЖР в монографиях ведущих зарубежных фармакопей, как правило, различается либо качественно, либо количественно и зависит от типа используемого масла, а также различаются требования к содержанию примесей в МЖР в зависимости от предполагаемой к производству лекарственной формы. В ГФ РФ отсутствуют частные фармакопейные статьи на наиболее часто используемые в процессе производства ЛП МЖР (подсолнечное, оливковое, кунжутное, рапсовое). Оценка качества таких масел осуществляется в соответствии с требованиями общей фармакопейной статьи (ОФС) «Масла жирные растительные» ГФ РФ. Требования же к оценке качества ЛП, представляющих собой масляные растворы, приведены только в ОФС «Растворы» ГФ РФ, согласно которой определяют «Кислотное число» и «Пероксидное число».
ВЫВОДЫ. При подготовке фармакопейных статей на ЛП, представляющие собой масляные растворы, необходимо стандартизировать подходы к оценке примесей, а именно предусмотреть обязательный контроль по показателям «Кислотное число» и «Пероксидное число», поскольку данные показатели характеризуют качество используемого масла как на момент производства, так и в процессе хранения.
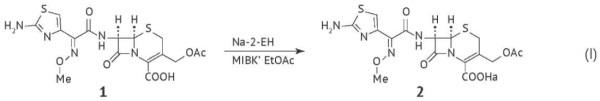
ВВЕДЕНИЕ. Отдельным классом органических примесей в фармацевтических субстанциях антибиотиков являются технологические примеси, не относящиеся к родственным соединениям или органическим растворителям, в частности N,N-диметиланилин, 2-этилгексановая кислота и ее производные. Данные вещества используются в процессе производства фармацевтических субстанций антибиотиков в качестве реагентов, и контроль их остаточного содержания предусмотрен отечественной и зарубежными фармакопеями.
ЦЕЛЬ. Обосновать требования к контролю технологических неродственных примесей в фармацевтических субстанциях антибиотиков.
МАТЕРИАЛЫ И МЕТОДЫ. Проведен анализ фармакопейных статей и монографий Государственной фармакопеи Российской Федерации (ГФ РФ) и зарубежных фармакопей (Европейской фармакопеи (Ph. Eur.), Фармакопеи США (USP), Международной фармакопеи) на фармацевтические субстанции антибиотиков, а также анализ материалов регистрационных досье на фармацевтические субстанции антибиотиков российского и зарубежного производств, включенных в Государственный реестр лекарственных средств. В работе использовали методы сравнительного информационно-аналитического исследования и контент-анализа.
РЕЗУЛЬТАТЫ. В Ph. Eur. контроль содержания 2-этилгексановой кислоты предусмотрен в фармацевтических субстанциях антибиотиков (полусинтетических и полученных способом ферментации). В USP этот показатель не включен в частные монографии. Определение остаточных количеств N,N-диметиланилина введено как в частные монографии на полусинтетические антибиотики Ph. Eur., так и USP, что, вероятно, обусловлено его высокой токсичностью. Требования ГФ РФ гармонизированы с Ph. Eur. При анализе материалов регистрационных досье было показано, что производители заменяют 2-этилгексановую кислоту и N,N-диметиланилин на менее токсичные реагенты, содержание которых при необходимости контролируют в соответствии с общими требованиями к остаточным органическим растворителям.
ВЫВОДЫ. Современные технологические процессы позволяют производить фармацевтические субстанции антибиотиков без использования 2-этилгексановой кислоты и N,N-диметиланилина. Концепция их контроля должна быть основана на оценке рисков, и соответствующие указания следует включить в фармакопейные статьи, переведя показатели из статуса обязательных в статус контролируемых только при использовании 2-этилгексановой кислоты и N,N-диметиланилина в процессе производства.
ВВЕДЕНИЕ. Одним из показателей качества лекарственных средств (ЛС) и вспомогательных веществ является содержание остаточных органических растворителей (ООР). В ОФС.1.1.0008 «Остаточные органические растворители» Государственной фармакопеи Российской Федерации (ГФ РФ) отсутствуют методики идентификации, установления предельного содержания и количественного определения ООР, что не позволяет стандартизировать подходы к контролю качества по этому показателю.
ЦЕЛЬ. Анализ мировой фармакопейной практики и методологических подходов к контролю ООР для подготовки проекта общей фармакопейной статьи «Остаточные органические растворители».
ОБСУЖДЕНИЕ. В результате сравнительного анализа требований Европейской фармакопеи, Фармакопеи США, Фармакопеи Евразийского экономического союза и ГФ РФ к контролю ООР отмечено отсутствие описания хроматографических систем в действующей ОФС ГФ РФ и установлена необходимость их включения. Отмечено, что ни в одной из рассмотренных фармакопей требования к валидации соответствующих аналитических методик не устанавливают исчерпывающий перечень характеристик оборудования, необходимый и достаточный для воспроизведения всех условий выполнения анализа.
ВЫВОДЫ. Целесообразно гармонизировать ОФС «Остаточные органические растворители» ГФ РФ в соответствии с мировыми фармакопейными подходами к определению ООР методом газовой хроматографии. Рекомендовано дополнить проект ОФС «Остаточные органические растворители»» описанием методик идентификации, установления предельного содержания и количественного определения ООР.
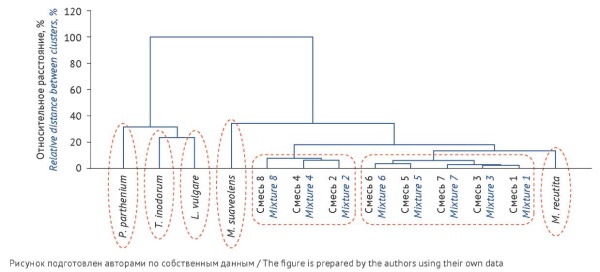
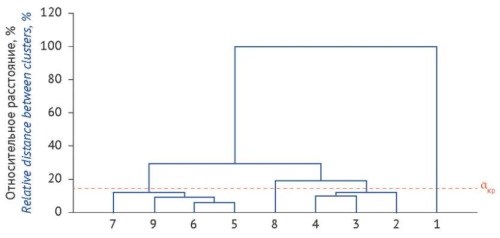
ВВЕДЕНИЕ. Определение подлинности лекарственного растительного сырья и его отличий от морфологически схожих видов часто оказывается затруднительным, особенно при использовании измельченного сырья. Для анализа сложных смесей растительного происхождения целесообразно использовать дополнительные способы оценки подлинности, в частности метод иерархической кластеризации по содержанию микроэлементов в сырье.
ЦЕЛЬ. Исследовать возможность использования метода иерархической кластеризации содержания микроэлементов в сложной биологической матрице (смеси растений) на примере ромашки аптечной.
МАТЕРИАЛЫ И МЕТОДЫ. Для ромашки аптечной, широко применяемой в медицинской практике, известен ряд морфологически схожих совместно произрастающих видов других растений. Объектами исследования являлись цветки с цветоносами, заготовленные от растений: Matricaria recutita L., Tanacetum parthenium (L.) Sch.Bip., Leucanthemum vulgare Lam., Tripleurospermum inodorum (L.) Sch.Bip. и Matricaria suaveolens Buchenau и искусственно созданные смеси ромашки и примесных видов, не соответствующие требованиям фармакопейной статьи. Содержание микроэлементов определяли методом масс-спектрометрии с индуктивно-связанной плазмой. Статистическую обработку результатов проводили с помощью программы Statistica 10 со встроенными алгоритмами анализа данных многофакторных экспериментов.
РЕЗУЛЬТАТЫ. Определено содержание 56 микроэлементов в растениях и их смесях, затем с использованием методов кластерного анализа построено иерархическое дерево. Выявлено, что наиболее близкими по микроэлементному статусу являются M. recutita и M. suaveolens, что коррелирует с их таксономией. Искусственно приготовленные смеси растений занимали промежуточные кластеры между различными видами ромашки. При этом по критерию несходства все примесные виды растений и смеси с содержанием более 10% значимо отличались от кластера микроэлементного состава M. recutita.
ВЫВОДЫ. Показана возможность использования метода иерархической кластеризации содержания микроэлементов в биологическом объекте для анализа сложных систем, в том числе смесей растений, и выявления их различий, способных иметь диагностическое значение при контроле качества лекарственного растительного сырья.
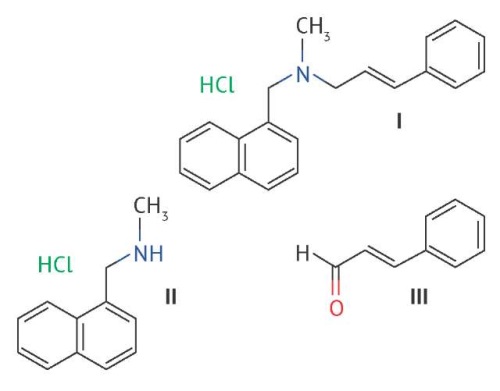
ВВЕДЕНИЕ. Лекарственные препараты на основе нафтифина широко используются в медицинской практике для лечения грибковых инфекций. Одним из основных показателей качества лекарственного препарата является содержание действующего вещества. При разработке методик количественного определения для рутинного анализа лекарственных средств особое внимание уделяется сокращению временных и материальных затрат.
ЦЕЛЬ. Модернизация методики количественного определения нафтифина и его примесей в лекарственных препаратах методом ВЭЖХ с использованием колонок малого объема, позволяющих сократить время анализа и расход реактивов.
МАТЕРИАЛЫ И МЕТОДЫ. Объектами исследования были субстанция и препараты нафтифина в форме 1% спиртового раствора и крема для наружного применения. Хроматографирование растворов проводили на жидкостных хроматографах Agilent 1200 Infinity и Agilent Infinity II 1290, оснащенных диодно-матричными детекторами с использованием нескольких хроматографических колонок: XBridge Phenyl 20×4,6 мм с размерами частиц 2,5 и 3,5 мкм и Acquity BEH Phenyl 75×2,1 мм, 1,7 мкм. Для оценки специфичности методики использовали образцы N-метил-1-нафталинметиламина, коричного альдегида и растворы нафтифина после химической, термической и фотолитической деструкции.
РЕЗУЛЬТАТЫ. Подобран оптимальный нетоксичный растворитель проб — 0,1% раствор ортофосфорной кислоты и доказано, что для разных лекарственных форм могут быть применены разные растворители. Выбраны условия анализа: концентрация испытуемых растворов нафтифина — 10 мкг/мл, колонка XBridge Phenyl (20×4,6 мм; 2,5 мкм), градиентный режим элюирования смесью 0,1% раствора хлорной кислоты и ацетонитрила со скоростью 1 мл/мин. Показано, что детектирование при длине волны 254 нм обеспечивает наилучшее соотношение сигнала и шума для пика нафтифина. Воспроизводимость разработанной методики количественного определения нафтифина была подтверждена валидацией согласно требованиям Государственной фармакопеи Российской Федерации. Специфичность методики подтверждена хроматографированием растворителя, подвижной фазы и модельных растворов, содержащих основные примеси нафтифина. Линейность методики подтверждена в диапазоне 80–120% нафтифина (коэффициент корреляции составил 0,995). При проверке правильности методики открываемость составила 100,2%. Доказана устойчивость методики при незначительных изменениях хроматографических параметров. Время удерживания пика нафтифина — около 2 мин.
ВЫВОДЫ. Разработана селективная и чувствительная ВЭЖХ-методика количественного определения нафтифина в лекарственных средствах, позволяющая сократить время анализа и свести к минимуму расход используемых реагентов. Результаты валидации методики удовлетворяют критериям приемлемости, подтверждают ее пригодность и воспроизводимость.
ЛЕКАРСТВЕННЫЕ СРЕДСТВА РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
ВВЕДЕНИЕ. Грибы рода Ganoderma spp., используемые в восточной медицине и в качестве биологически активных добавок к пище, представляют интерес как источники эффективных антиоксидантов. В Российской Федерации и Республике Беларусь не разработана нормативная документация по контролю качества этого вида сырья и его биологически активных веществ, поэтому исследование химического состава и спектра фармакологической активности экстрактов G. lingzhi и G. lucidum является актуальной задачей.
ЦЕЛЬ. Установление химического состава и антиоксидантной активности экстрактов плодовых тел G. lingzhi и G. lucidum.
МАТЕРИАЛЫ И МЕТОДЫ. Объектами исследования явились чистые культуры G. lingzhi и G. lucidum из коллекции штаммов грибов ГНУ «Институт леса НАН Беларуси». Для культивирования биомассы использовали два субстрата на основе опилок ольхи (степень измельчения 1–3 мм) и стружки дуба (степень измельчения 5–10 мм). Экстрагирование биомассы проводили 70% этанолом по методу ремацерации. Для определения антирадикальной активности экстрактов грибов использовали реакцию со стабильным свободным радикалом DPPH (2,2-дифенил-1-пикрилгидразил) и катион-радикалом ABTS (2,2’-азино-бис(3-этилбензтиазолин-6-сульфоновой кислоты). Химический состав экстрактов исследовали методом хроматомасс-спектрометрии. Определение количества фенольных, стероидных и тритерпеновых соединений проводили спектрофотометрически.
РЕЗУЛЬТАТЫ. Установлено, что наибольшую антирадикальную активность проявляет экстракт штамма 334 G. lucidum, выращенный на субстрате ольхи (концентрация полумаксимального ингибирования (IC50) по реакции с DPPH=3,1±0,2 мкг/мл, IC50 (ABTS)=3,7±0,2 мкг/мл) и характеризующийся максимальным содержанием фенольных (326,2±16,5 мкмоль/г) и тритерпеновых соединений (2,00±0,11 ммоль/г). Антиоксидантная активность полученных экстрактов может быть обусловлена содержанием ганодеровой кислоты D, люциденовой кислоты D и нарингенина и других фенольных соединений.
ВЫВОДЫ. Высокий выход экстракта со значительным уровнем антиоксидантной активности позволяет рассматривать искусственно выращенные грибы G. lingzhi и G. lucidum в качестве перспективных источников природных антиоксидантов.
ВВЕДЕНИЕ. Фармакопейная методика определения содержания тяжелых металлов и мышьяка в лекарственном растительном сырье не позволяет различить элементные примеси эндогенного и экзогенного происхождения. При этом более точные данные о микроэлементном составе растения, в том числе информация об эндо- и экзогенных примесях, могли бы быть использованы в задачах хемосистематики и для контроля подлинности сырья.
ЦЕЛЬ. Модификация методики определения содержания макро- и микроэлементов в растительном сырье для исключения влияния экзогенного загрязнения на результаты анализа.
МАТЕРИАЛЫ И МЕТОДЫ. В качестве объектов исследования были выбраны растения семейства бурачниковых: генеративные побеги медуницы мягкой — Pulmonaria mollis Wulf. ex Hornem и листья бурачника лекарственного — Borago officinalis L. В часть сырья вносили фиксированное количество элементов-поллютантов. Все сырье измельчали и разделяли на фракции. Количественное определение проводили методом масс-спектрометрии с индуктивно-связанной плазмой. Статистическую обработку результатов проводили с использованием программ Microsoft Excel и Statistica 8.
РЕЗУЛЬТАТЫ. В 9 фракциях сырья с различными размерами частиц было определено содержание 30 элементов. Кластерный анализ показал, что фракция сырья с размерами частиц менее 0,2 мм и нефракционированное сырье образуют отдельные кластеры, что свидетельствует об их значимо разном элементном составе. Статистический анализ с использованием критерия Граббса позволил подтвердить, что эти выборки значимо отличались от остальных. Содержание элементов-поллютантов, введенных в фиксированном количестве в сырье, оказалось максимально во фракции частиц менее 0,2 мм — более 90% от введенного количества. Высокое содержание элементов в нефракционированном сырье являлось следствием присутствия частей растения, которые при измельчении и фракционировании попадают во фракцию с размером частиц менее 0,2 мм.
ВЫВОДЫ. Использование нефракционированного сырья при определении содержания элементов вносит систематическую погрешность в результаты измерений и затрудняет их использование для контроля подлинности сырья. Для получения достоверных результатов определения микроэлементного статуса растений предложено при пробоподготовке проводить дополнительное фракционирование сырья и не использовать для анализа фракцию с размерами частиц менее 0,2 мм.
ФАРМАЦЕВТИЧЕСКОЕ ПРОИЗВОДСТВО
ВВЕДЕНИЕ. Актуальной задачей фармацевтической отрасли является разработка ресурсосберегающих технологий производства лекарственных средств (ЛРС). Один из способов эффективного использования лекарственного растительного сырья — технологии, позволяющие получать продукты с различным фармакологическим действием в одном технологическом цикле. В настоящее время отсутствует регламентированный способ обработки данного вида сырья, который бы позволил разрабатывать лекарственные препараты на основе календулы цветков с заданным фармакологическим действием. В странах Евразийского экономического союза (ЕАЭС) для переработки сырья календулы используются методы экстракции, направленные на получение в одном технологическом цикле только одной фракции БАВ (только флавоноидов или только каротиноидов).
ЦЕЛЬ. Оценка возможности получения фракций биологически активных веществ разной полярности (каротиноиды, флавоноиды и полисахариды) из календулы цветков путем использования поэтапной обработки лекарственного растительного сырья в одном технологическом цикле.
МАТЕРИАЛЫ И МЕТОДЫ. Экстракцию каротиноидов календулы цветков проводили гексаном. Дополнительно для предварительной обработки сырья использовали термическое воздействие. Количественное определение каротиноидов и флавоноидов выполняли спектрофотометрическим методом, полисахаридных фракций — гравиметрическим. После отгонки гексана был получен твердый маслянистый остаток, который растворяли при механическом перемешивании в масле для получения масляного экстракта. Выполняли трехэтапную обработку: экстракция гексаном (экстракция слабополярным органическим растворителем каротиноидов), экстракция смесью вода/ацетон в отношении 30/70 по объему (выделение флавоноидов) и экстракция водой (осаждение полисахаридов).
РЕЗУЛЬТАТЫ. Максимальное количество каротиноидов (~4%) содержится в липофильных вытяжках, полученных при однократной экстракции гексаном и ее комбинации с термообработкой, при водно-органической экстракции их содержание меньше в десять раз. Масляные экстракты были получены из твердого маслянистого остатка после отгонки гексана при температуре его кипения, содержание биологически активных веществ в них сопоставимо с исходным. Предварительная экстракция гексаном увеличивает выход флавоноидов на 68,5% при водно-органической экстракции и снижает его в 1,7 раза при водной экстракции по сравнению с нативным сырьем, что сопровождается уменьшением их содержания во фракции водорастворимых полисахаридов практически в 10 раз. С введением новых этапов обработки суммарное содержание и содержание отдельных полисахаридных фракций снижается с повышением степени их чистоты. Соотношение полисахаридных фракций не изменяется при использовании всех примененных способов обработки.
ВЫВОДЫ. Поэтапная обработка сырья календулы цветков позволяет в одном технологическом цикле выделить три фракции, содержащие повышенное количество каротиноидов (в 3,3 раза), флавоноидов (на 44,8%) и полисахаридов (на 41,3%) соответственно в сравнении с нативным сырьем. Полученный продукт характеризуется более высокой степенью чистоты, чем продукт, полученный при предобработке с применением двукратной экстракции гексаном, или из нативного сырья. Метод рекомендовано использовать для получения лекарственных средств, обогащенных конкретной группой БАВ, или с меньшим содержанием примесей.
ДОКЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ

ВВЕДЕНИЕ. Терапия стволовыми клетками представляет собой перспективный метод лечения различных заболеваний и травм, однако безопасность этого метода изучена недостаточно. Исследование безопасности ретроорбитального введения ксеногенного биомедицинского клеточного продукта необходимо для разработки протоколов дальнейших исследований потенциальных препаратов при терапии неврологических заболеваний.
ЦЕЛЬ. Выбор дозы биомедицинского клеточного продукта, полученного на основе глиальных клеток-предшественников, и оценка его безопасности при ретроорбитальном введении мышам линии C57BL/6J.
МАТЕРИАЛЫ И МЕТОДЫ. Глиальные клетки-предшественники (ГКП), полученные из индуцированных плюрипотентных стволовых клеток человека путем их поэтапной дифференцировки, культивировали в среде DMEM/F12 с добавлением эпидермального фактора роста и цилиарного нейротрофического фактора, в качестве подложки использовали матригель. Введение клеток мышам осуществляли ретроорбитально под изофлурановым наркозом один раз в неделю на протяжении двух месяцев. Исследование проводили на самцах мышей линии C57BL/6J. Оценивали биохимические показатели крови животных, изменения в поведении, количество активированных астроцитов и клеток микроглии методом иммуногистохимического анализа.
РЕЗУЛЬТАТЫ. При введении ГКП в дозе 500×10³ кл./мышь, выбранной на основании данных литературы, в плазме крови животных было отмечено увеличение концентрации аланинаминотрансферазы и аспартатаминотранферазы, что могло свидетельствовать о повреждении клеток и развитии воспалительных реакций. При уменьшении дозы вводимых клеток в 3 раза и более биохимические показатели крови не отличались от результатов в группе контроля. При оценке маркеров нейровоспаления в экспериментах с разными дозами ГКП значимых различий выявлено не было, однако у животных, получавших препарат в дозе 150×10³ кл./мышь, наблюдали увеличение числа астроцитов, что может свидетельствовать о развивающихся воспалительных процессах в головном мозге. При введении ГКП в дозах 50×10³ и 15×10³ кл./мышь в ретроорбитальный венозный синус патологических изменений в головном мозге и в плазме крови животных не наблюдалось.
ВЫВОДЫ. Полученные результаты свидетельствуют о потенциальной безопасности длительной терапии ГКП на мышах при соблюдении оптимальных условий дозирования. Установленные оптимальные дозы и способ введения ГКП предложено использовать для дальнейших исследований безопасности внутривенной доставки клеточных продуктов, предназначенных для терапии неврологических заболеваний.

ISSN 3034-3453 (Online)





























