


ГЛАВНАЯ ТЕМА: ГАРМОНИЗАЦИЯ ФАРМАКОПЕЙНЫХ СТАНДАРТОВ: ОПЫТ РОССИЙСКОЙ ФЕДЕРАЦИИ

В интервью рассматриваются ключевые аспекты гармонизации фармакопейных стандартов на лекарственные средства в условиях глобализации фармацевтического рынка. Автор анализирует опыт международных организаций (ВОЗ, ICH, PDG) и оценивает реалистичность создания единых мировых стандартов качества. Особое внимание уделено процессу формирования Фармакопеи ЕАЭС, принципам унификации терминологии, разработке Номенклатуры лекарственных форм, а также методологическим различиям между ведущими фармакопеями (USP, Ph. Eur., BP, JP). Выявлены основные барьеры на пути к полной унификации: расхождения в подходах к идентификации, контролю примесей, нормированию содержания действующих веществ и проблеме взаимозаменяемости фармакопейных стандартных образцов. Делается вывод, что единый мировой стандарт качества остается долгосрочным ориентиром, тогда как региональная гармонизация в рамках ЕАЭС представляет собой эффективный и практичный этап сближения регуляторных требований.

ВВЕДЕНИЕ. Государственная фармакопея Российской Федерации ( ГФ РФ) является ключевым сводом обязательных требований к качеству лекарственных средств, при этом векторы ее развития не только основываются на совершенствовании правового поля, но также учитывают и основные тренды развития региональной фармакопеи — Фармакопеи Евразийского экономического союза (ЕАЭС). Необходимым условием для обеспечения устойчивого и последовательного совершенствования фармакопейных подходов и требований является проведение регулярного анализа актуального состояния фармакопеи и определение наиболее перспективных и востребованных направлений дальнейшего развития.
ЦЕЛЬ. Определение перспективных направлений развития Государственной фармакопеи Российской Федерации в условиях формирования единого фармацевтического рынка ЕАЭС.
ОБСУЖДЕНИЕ. Рассмотрены основные изменения в правовом поле на национальном и региональном (ЕАЭС) уровнях, в том числе обновленный Порядок разработки статей для ГФ РФ, содержащий прямое указание о необходимости учета требований Фармакопеи ЕАЭС при разработке российской фармакопеи, и Стратегия развития Фармакопеи ЕАЭС на период до 2035 г., содержащая базовые принципы, цель Стратегии, этапы ее реализации и необходимые мероприятия. Приведены актуальные сведения о составе ГФ РФ XV изд., а также предложены перспективные направления развития фармакопейных требований.
ВЫВОДЫ. С учетом основных трендов развития Фармакопеи ЕАЭС могут быть выделены следующие направления развития фармакопейных стандартов качества: актуализация номенклатуры и содержания общих фармакопейных статей на лекарственные формы лекарственных препаратов; распространение принципа гармонизации на сближение подходов с фармакопеями дружественных Российской Федерации государств; внедрение инструментальных, точных и селективных методов и методик, в том числе с целью замены испытаний с применением животных.
ВВЕДЕНИЕ. Лекарственные препараты (ЛП) лоперамида гидрохлорида зарегистрированы в РФ в лекарственных формах (ЛФ) «таблетки» и «капсулы». Однако в Государственную фармакопею Российской Федерации (ГФ РФ) не включены фармакопейные статьи на ЛФ лоперамида гидрохлорида. Представляется актуальным провести сравнительный анализ и обобщить требования зарубежных фармакопей к контролю качества ЛП на основе лоперамида гидрохлорида.
ЦЕЛЬ. Анализ и обобщение требований к качеству лекарственных средств лоперамида гидрохлорида для подготовки рекомендаций по составлению спецификаций на препараты, содержащие лоперамида гидрохлорид в ЛФ «таблетки» и «капсулы», и рекомендаций по актуализации действующей фармакопейной статьи на фармацевтическую субстанцию лоперамида гидрохлорида.
МАТЕРИАЛЫ И МЕТОДЫ. Проведено исследование требований ГФ РФ, Европейской фармакопеи (Ph. Eur.), Британской фармакопеи (ВР), Фармакопеи США (USP), Индийской фармакопеи (IP), Фармакопеи Китайской Народной Республики (ChP), Корейской фармакопеи (KP) и Международной фармакопеи (Ph. Int.) к качеству фармацевтических субстанций лоперамида гидрохлорида, препаратов лоперамида гидрохлорида в ЛФ «таблетки» и «капсулы». Рассмотрены материалы регистрационных досье на ЛС лоперамида гидрохлорида.
РЕЗУЛЬТАТЫ. ЛП лоперамида гидрохлорид не описан в ГФ РФ. Фармакопейная статья (монография) на фармацевтическую субстанцию лоперамида гидрохлорида включена в ГФ РФ XV изд., в Ph. Eur., ВР, USP, IP, ChP, КР и Ph. Int. Установлено, что ФС.2.1.0613 «Лоперамида гидрохлорид» ГФ РФ в значительной степени гармонизирована с Ph. Eur., ее требования являются одними из более строгих, и используются более совершенные методики идентификации и контроля примесей по сравнению с USP, IP, ChP и KP. Однако при пересмотре фармакопейных статей следует оптимизировать комбинацию методов идентификации лоперамида, исключить контроль тяжелых металлов полуколичественным методом, заменив его на оценку рисков содержания элементных примесей.
ВЫВОДЫ. При подготовке спецификаций на препараты лоперамида в ЛФ «таблетки» и «капсулы» даны рекомендации по использованию сочетаний методов идентификации, установлены возможности различий в условиях и нормах испытания на растворение, определены специфицируемые примеси. На примере ЛП лоперамида в различных модификациях показаны направления совершенствования стандартизации препаратов в ЛФ «таблетки» в части необходимости проведения испытаний как на распадаемость, так и на растворение. Обоснована возможность контроля однородности дозирования «таблеток-лиофилизатов» независимо от дозировки расчетно-массовым способом.
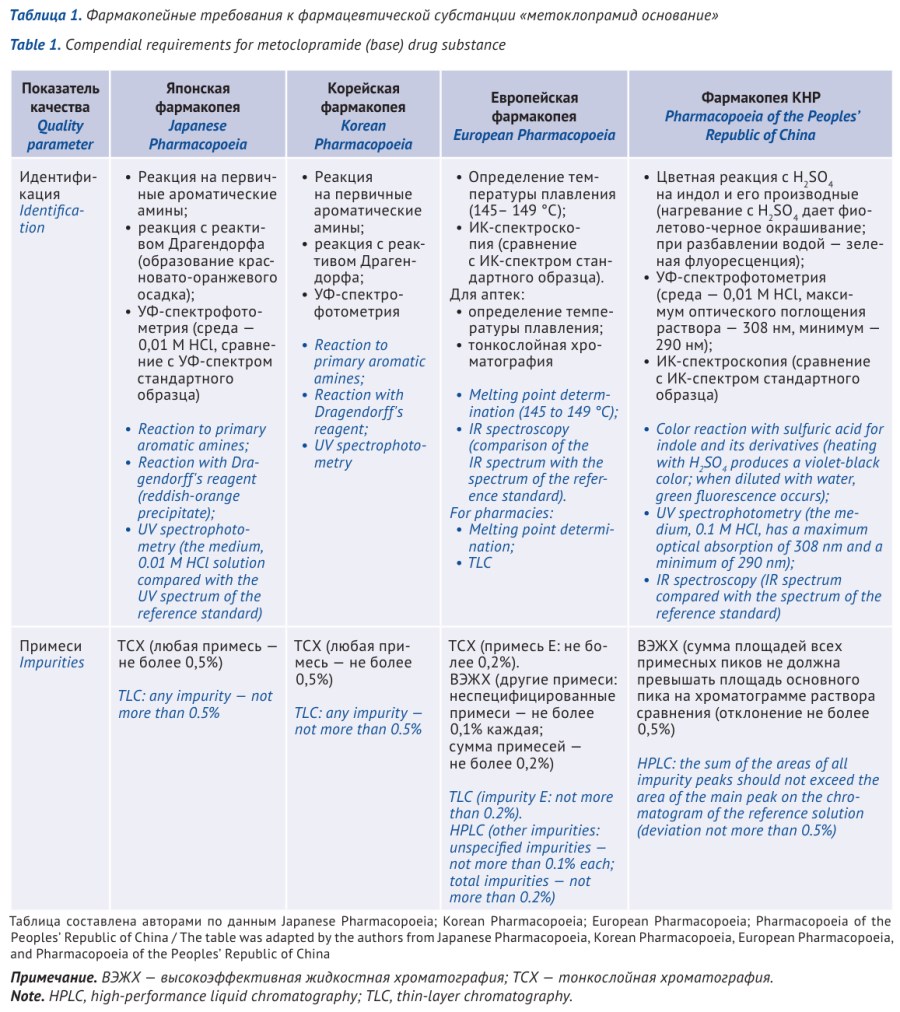
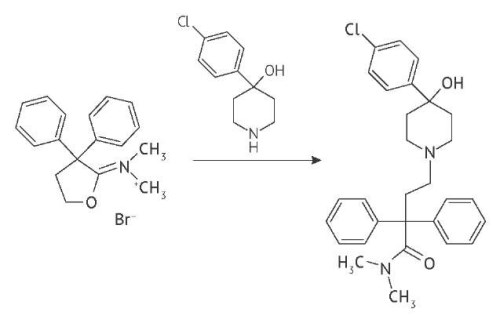
ВВЕДЕНИЕ. Метоклопрамид — лекарственный препарат, который широко применяется в качестве противорвотного средства. В фармацевтической промышленности используется метоклопрамид в форме основания или гидрохлорида моногидрата. Фармакопейная статья на фармацевтическую субстанцию «метоклопрамида гидрохлорид моногидрат» включена в Государственную фармакопею Российской Федерации (ГФ РФ), однако требования в качеству лекарственных препаратов на основе метоклопрамида в ГФ РФ отсутствуют. Для последующей разработки фармакопейной статьи на лекарственные препараты метоклопрамида представляется необходимой систематизация действующих зарубежных и российских требований к лекарственным средствам на основе метоклопрамида.
ЦЕЛЬ. Разработка подхода к контролю качества лекарственного препарата в лекарственной форме «таблетки» и фармацевтической субстанции метоклопрамид (гидрохлорид и основание).
МАТЕРИАЛЫ И МЕТОДЫ. В работе использовали методы сравнительного информационно-аналитического исследования и контент-анализа. Проведено исследование требований ГФ РФ XV изд. и ведущих зарубежных фармакопей: Европейской фармакопеи (Ph. Eur.), Британской фармакопеи (ВР), Фармакопеи США (USP), Индийской фармакопеи (IP), Фармакопеи Китайской Народной Республики (ChP), Японской фармакопеи (JP), Корейской фармакопеи (КР).
РЕЗУЛЬТАТЫ. Обоснован выбор показателей качества, методов анализа и критериев приемлемости для подготовки рекомендаций по составлению спецификаций на фармацевтическую субстанцию «метоклопрамид» основания и солевой формы, а также лекарственного препарата в лекарственной форме «таблетки». Проведен сравнительный анализ требований ГФ РФ и зарубежных фармакопей, а также изучены материалы регистрационных досье на фармацевтические субстанции и препараты в части ключевых показателей качества фармацевтической субстанции метоклопрамида: «Идентификация», «Родственные примеси»,
«Количественное определение» для субстанций и для таблеток; дополнительно для таблеток — «Растворение». В ходе проведенного анализа обоснован выбор аналитических методов контроля качества по показателю «Идентификация»: для фармацевтической субстанции — спектроскопия в ближней инфракрасной области, тонкослойная хроматография; для лекарственного препарата — высокоэффективная жидкостная хроматография с диодно-матричным детектором или комбинация методов высокоэффективной жидкостной хроматографии
и спектрофотометрии в ультрафиолетовой и видимой областях. Установлен допустимый уровень единичных примесей по показателю «Родственные примеси» для фармацевтической субстанции и препарата. Рекомендованы условия проведения испытаний по показателям качества «Растворение» и «Количественное определение». Даны рекомендации по включению показателей качества и методов анализа в фармакопейную статью на метоклопрамид основание. Предложено сокращение количества методов идентификации, указанных в ФС «Метоклопрамида гидрохлорид моногидрат».
ВЫВОДЫ. На основании сравнительного анализа фармакопейных требований предложен подход к формированию требований к качеству лекарственных средств метоклопрамида (субстанции и лекарственного препарата в форме «таблетки»). В качестве основы нормирования любой единичной примеси и суммы примесей предложено использовать подход Британской фармакопеи. Рекомендовано включить в ГФ РФ ФС на фармацевтическую субстанцию «метоклопрамид основание»
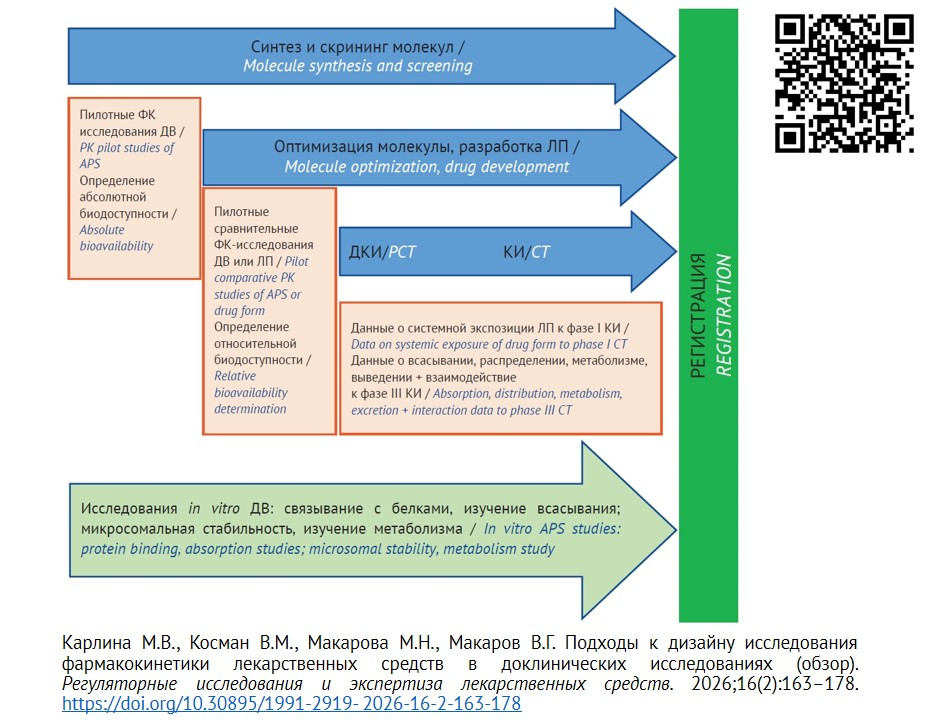
ВВЕДЕНИЕ. В соответствии с требованиями по процедуре регистрации лекарственных препаратов (ЛП) в регистрационное досье должны быть включены данные по доклиническому изучению фармакокинетики лекарственных средств. Однако вопрос объема предоставляемых данных для различных групп препаратов, а также определение того, какие фармакокинетические процессы и их параметры на каждом этапе жизненного цикла препарата должны быть изучены, остается открытым.
ЦЕЛЬ. Анализ данных литературы, отечественных и зарубежных методических документов по доклиническому изучению фармакокинетики лекарственных средств для выбора оптимальной стратегии сбора данных фармакокинетики на разных этапах жизненного цикла лекарственного препарата.
ОБСУЖДЕНИЕ. Материалы исследования — регуляторные документы, руководства по доклиническому изучению ЛП, научные статьи и иные литературные источники, находящиеся в открытом доступе (в том числе по данным электронных баз РИНЦ (eLIBRARY.RU), PubMed, Web of Science). Рассмотрены основные подходы к планированию исследований фармакокинетики для различных лекарственных препаратов (оригинальных, воспроизведенных, биологических и т.д.), включая выбор вида и количества лабораторных животных, исследуемых доз и временных точек отбора биоматериала.
ВЫВОДЫ. Исследования фармакокинетики лекарственных препаратов на этапе доклинических исследований необходимы для оптимизации структуры молекулы действующего вещества, выбора оптимального пути введения и оптимальной лекарственной формы, прогнозирования значений фармакокинетических параметров у человека, сокращения временных затрат и рисков при разработке безопасных и эффективных лекарственных средств. Предложены дизайны исследований фармакокинетики на этапе скрининга молекул, оптимизации молекул и выбора лекарственной формы для исследований различных групп препаратов.
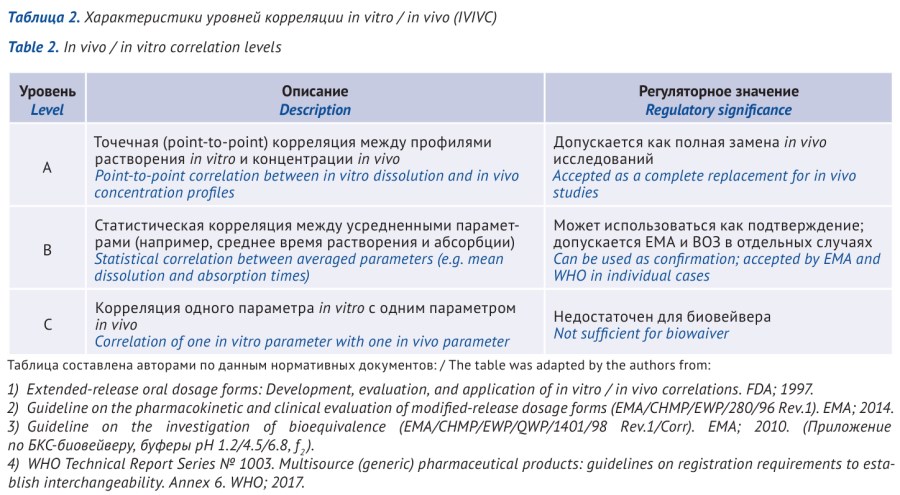
ВВЕДЕНИЕ. Процедура биовейвер на основе биофармацевтической классификационной системы (БКС) широко применяется в регуляторной практике при оценке биоэквивалентности воспроизведенных лекарственных препаратов (ВЛП). Применение процедуры биовейвер направлено на сокращение объема исследований in vivo при сохранении требований к качеству, безопасности и эффективности ВЛП. В последние годы в научной литературе активно обсуждаются дополнительные аналитические и модельные инструменты, используемые при оценке биоэквивалентности. Это требует четкого разграничения между регуляторными требованиями и исследовательскими подходами, включая корреляции in vitro / in vivo (IVIVC), физиологически обоснованное фармакокинетическое моделирование (PBPK), а также использование алгоритмов искусственного интеллекта.
ЦЕЛЬ. Сравнительный анализ современных нормативных подходов к применению процедуры биовейвер на основе биофармацевтической классификационной системы, а также систематизация действующих требований, применяемых при оценке биоэквивалентности воспроизведенных лекарственных препаратов.
ОБСУЖДЕНИЕ. Во всех рассматриваемых регуляторных системах применение процедуры биовейвер нормативно ограничено активными фармацевтическими ингредиентами классов I и III БКС при строгом соблюдении критериев растворимости и сопоставимости профилей растворения. Деление класса II на подклассы IIa и IIb не закреплено ни в одном из официальных регуляторных документов и используется исключительно в научной литературе в исследовательском контексте. Установлено, что такие модели, как IVIVC, PBPK и технологии искусственного интеллекта, рассматриваются регулирующими органами как вспомогательные научные инструменты и не имеют самостоятельного статуса доказательной базы для отказа от исследований in vivo.
ВЫВОДЫ. Результаты обзора подтверждают отсутствие принципиальных регуляторных расхождений между подходами к применению процедуры биовейвер. Выявленные в литературе предложения по расширению ее применения относятся к научной дискуссии и не отражают действующую регуляторную практику. Работа формирует методологически корректную основу для интерпретации регуляторных требований и дальнейших исследований в области биофармацевтической оценки ВЛП.
ВВЕДЕНИЕ. Ванкомицин — трициклический гликопептидный антибиотик, продуцируемый Amycolatopsis orientalis. Для определения активности антибиотиков, получаемых путем биосинтеза, в частности ванкомицина, для которых не предусмотрены химические или физико-химические методы определения активного соединения, применяют два фармакопейных метода: метод диффузии в агар (МДА) и турбидиметрический метод (ТМ). Оба метода основаны на угнетении роста тест-микроорганизма в соответствующих условиях эксперимента. Для оценки активности парентеральных препаратов, применяемых в виде растворов, турбидиметрический метод имеет ряд преимуществ по сравнению с методом диффузии в агар как более чувствительный к низким концентрациям, более экономичный и не требующий длительного периода времени для получения результатов. Государственной фармакопеей Российской Федерации предусмотрен один метод определения активности антибиотиков — метод диффузии в агар. Ранее была разработана методика определения активности ванкомицина турбидиметрическим методом.
ЦЕЛЬ. Оценка результатов определения активности ванкомицина, полученных турбидиметрическим методом, с целью использования его в качестве альтернативного методу диффузии в агар.
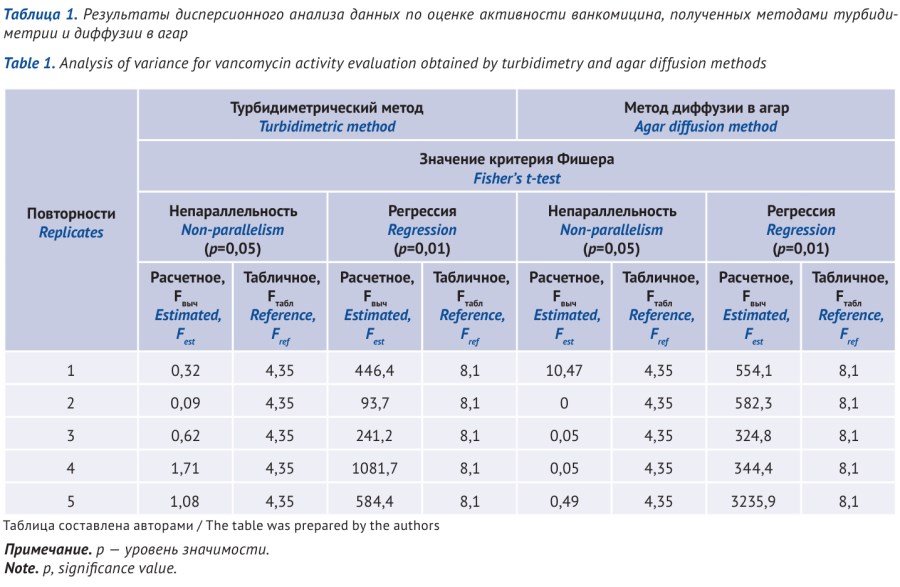
МАТЕРИАЛЫ И МЕТОДЫ. Условия проведения испытания методики с использованием МДА соответствовали условиям, приведенным в Государственной фармакопее Российской Федерации: тест-микроорганизм, посевная доза: Васillus subtilis NCTC 10400/АТСС 6633 — Becton Dickinson France S.A.S., 20 млн спор на 1 мл среды, для приготовления основных растворов — стерильная вода очищенная, растворитель для рабочих растворов — стерильный буфер № 4. Методика ТМ теста — микроорганизм Staphylococcus aureus NCTC 7447/АТСС 6538P — Becton Dickinson France S.A.S., растворитель для приготовления — стерильная вода очищенная, растворитель для приготовления рабочих растворов — стерильный буферный раствор с рН 8,0 (заимствовано из Европейской фармакопеи). Использовали питательную среду, разработанную в ФГБУ «НЦЭСМП» Минздрава России для определения активности аминогликозидов. Определение активности проводили сравнением со стандартным образцом ванкомицина гидрохлорида EP CRS banth 4 для микробиологических анализов. Для испытания по каждому методу (МДА и ТМ) отбирали по 5 навесок с точностью до 0,0001 г испытуемого образца, каждую навеску анализировали в трех повторностях.
РЕЗУЛЬТАТЫ. Расчет основных параметров дисперсионного анализа «непараллельность» и «регрессия» результатов испытуемых и стандартных растворов подтвердил равноценность данных, полученных обоими методами. Сравнение выборочных дисперсий анализа двух методик (ТМ и МДА) по критерию Фишера и по критерию Бартлетта, значению χ2 при уровне вероятности Р=95% и f=1 и сравнение средних результатов по t-критерию Стьюдента подтвердили принадлежность обеих выборок к одной генеральной совокупности. Систематическая ошибка (ε) и неопределенность (δ) методики ТМ существенно выше, чем при применении методики МДА: ε=1,5 и 0,77%; δ=4,7 и 0,97% соответственно.
ВЫВОДЫ. Турбидиметрический метод может рассматриваться в качестве альтернативного метода определения активности ванкомицина. Однако для минимизации влияния человеческого фактора и внешних условий испытания необходимо предварительно разработать стандартные операционные процедуры выполнения работ по методике ТМ и контролировать их соблюдение.
ВВЕДЕНИЕ. Плоды тмина обыкновенного применяются для лечения синдрома раздраженного кишечника и диспептических расстройств благодаря их выраженному спазмолитическому, противомикробному и ветрогонному действию. Спазмолитическая активность плодов тмина обусловлена преимущественно содержанием эфирного масла, преобладающим компонентом которого является карвон. Европейской фармакопеей 11 изд. регламентируется содержание карвона в эфирном масле, тогда как в Государственной фармакопее Российской Федерации XIV изд. стандартизация плодов тмина по содержанию карвона не предусмотрена.
ЦЕЛЬ. Разработка и валидация спектрофотометрической методики определения карвона в эфирном масле плодов тмина обыкновенного; проверка результатов спектрофотометрического определения методом газовой хроматографии — масс-спектрометрии.
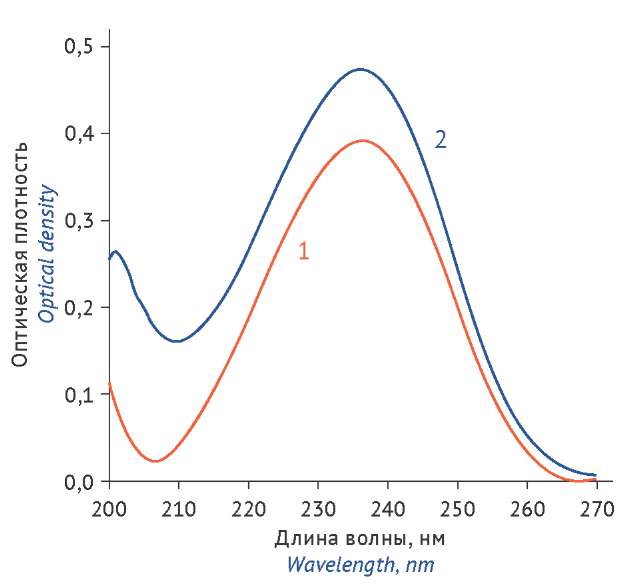
МАТЕРИАЛЫ И МЕТОДЫ. В работе использовали плоды тмина обыкновенного (Carum carvi L.) производства ООО «Фирма Здоровье» серия 031224, срок годности до 11.2027, стандартный образец карвона с содержанием основного вещества 98,5% (Fluka), спектрофотометр UNICO 2802 (United Products & Instruments, Inc.), весы аналитические АДВ-200М (ГОСМЕТР), газовый хроматограф GC-MS QP2010 Ultra (Shimadzu), совмещенный с масс-селективным детектором. При спектрофотометрическом определении навески эфирного масла и стандартного образца карвона растворяли в 95% спирте и снимали оптические плотности растворов при длине волны 236 нм в кюветах с толщиной слоя 10 мм. Результаты спектрофотометрической методики подтверждали с помощью газовой хроматографии.
РЕЗУЛЬТАТЫ. Валидация спектрофотометрической методики количественного определения карвона, проведенная в соответствии с требованиями ОФС.1.1.0012 «Валидация аналитических методик» Государственной фармакопеи Российской Федерации XV изд., показала ее специфичность, линейность, прецизионность и правильность. Результаты не отягощены систематической погрешностью. Относительное стандартное отклонение (RSD) не превышает 2,0%. Результаты спектрофотометрического определения карвона (75,7±0,9%) коррелируют с результатами анализа методом газожидкостной хроматографии (75,2±0,8%).
ВЫВОДЫ. Разработанная методика количественного определения карвона может быть использована для стандартизации как самих плодов тмина обыкновенного путем введения дополнительного критерия оценки качества его эфирного масла в раздел «Количественное определение», так и эфирного масла тмина обыкновенного, используемого в качестве субстанции для изготовления лекарственных препаратов.
ВВЕДЕНИЕ. Содержание пестицидов в лекарственном растительном сырье по российскому законодательству регламентируется ОФС.1.5.3.0011 «Определение содержания остаточных пестицидов в лекарственном растительном сырье и лекарственных растительных препаратах» Государственной фармакопеи Российской Федерации XV изд. (ГФ РФ). Остаточные количества пестицидов — один из показателей безопасности лекарственного растительного сырья. В ГФ РФ даны нормы по содержанию 65 подобных соединений, однако рынок ядохимикатов активно расширяется (рост составляет не менее 10% в год), и не все использующиеся соединения подлежат контролю при установлении качества продукции. Особый интерес представляют ядохимикаты, которые интенсивно применяются в сельском хозяйстве для уничтожения сорной растительности, — гербициды, содержащие, в частности, производное хлорбензойной кислоты дикамбу (3,6-дихлор-2-метоксибензойную кислоту). Дикамба используется в составе препаратов для борьбы с сорной растительностью «Линтур» и «Диален Супер».
ЦЕЛЬ. Разработка селективной методики определения гербицида дикамба в лекарственном растительном сырье эхинацеи пурпурной для последующего введения в фармакопейные нормы.
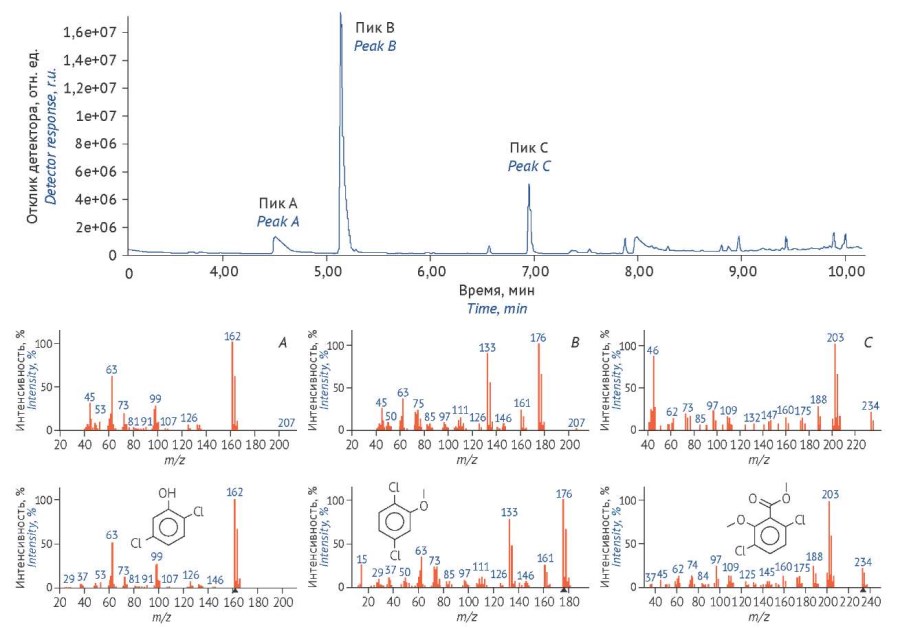
МАТЕРИАЛЫ И МЕТОДЫ. Стандартный образец дикамбы (Kit-54, pesticide 4-4600, PolуScience Corporation), растительный объект — эхинацея пурпурная сорта «Ливадия», выращенная из семян (срок годности до 06.2028, ООО «Аэлита»), гербицидная смесь — «Линтур», содержащая дикамбы — 659 г/кг, триасульфурона — 41 г/кг (ООО «Зеленая Аптека Садовода»). Исследования методом высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией (ВЭЖХ-МС/МС) проводили на модульном жидкостном хроматографе Agilent Technologies 1260 Infinity, детектор — тандемный масс-спектрометр Agilent Technologies 6420 Triple Quad LS/MS, исследования методом газовой хроматографии с масс-спектрометрическим детектором (ГХ-МС) проводили на газовом хроматографе, детектор — моноквадрупольный масс-спектрометр QP-2020 (Shimadzu). Хроматограммы и масс-спектры, полученные в ВЭЖХ-МС/МС исследованиях, анализировали в базах данных Agilent MassHunter Quantitative Analysis B.06.00, хроматограммы ГХ-МС исследования анализировали в программе Аgilent G1710 MSD Data Analysis ChemStation. Масс-спектры, полученные методом ГХ-МС, анализировали по библиотекам NIST MS Search Program 17.
РЕЗУЛЬТАТЫ. Разработана методика количественного определения гербицида дикамба в лекарственном растительном сырье методом ВЭЖХ-МС/МС, проведена оценка валидационных параметров (пригодность хроматографической системы, специфичность, линейность, правильность, прецизионность), изучена возможность применения метода ГХ-МС без/с дериватизацией силилированием. Рассчитано предельно допустимое значение содержания гербицида дикамба в лекарственном растительном сырье, сделан вывод о несоответствии сырья эхинацеи пурпурной, выращенной с применением данного гербицида, фармакопейным требованиям.
ВЫВОДЫ. Показана пригодность методики ВЭЖХ-МС/МС и непригодность метода ГХ-МС для количественного определения дикамбы в лекарственном растительном сырье. Разработанная методика может быть предложена в качестве основной для определения дикамбы в лекарственном растительном сырье «трава» для включения в фармакопейные требования.
ВВЕДЕНИЕ. Лекарственные средства, получаемые с использованием технологии рекомбинантной ДНК, широко применяются в терапии онкологических, иммуновоспалительных и инфекционных заболеваний. Объем исследований по разработке новых белковых препаратов, в том числе моноклональных антител, а также методик контроля качества таких препаратов увеличивается. Пептидное картирование позволяет подтвердить подлинность, первичную структуру белка, генетическую стабильность и идентифицировать изменения в структуре.
ЦЕЛЬ. Систематизация современных методологических подходов к разработке методик пептидного картирования.
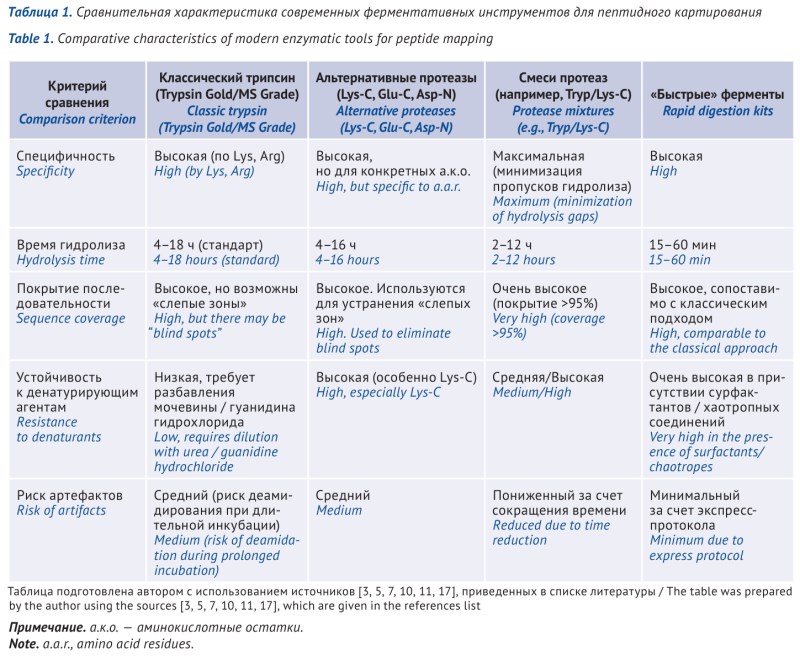
ОБСУЖДЕНИЕ. Одним из главных методов подтверждения подлинности белков является пептидное картирование, основанное на ферментативном гидролизе белка, с получением уникального набора пептидных фрагментов. Несмотря на уникальность каждой конкретной методики, все они базируются на общих принципах пробоподготовки и анализа, а также регуляторных требованиях. Разработка метода — сложный многостадийный процесс. В настоящее время для расщепления белка используются различные ферменты, однако «золотым
стандартом» является трипсин. Все большее распространение имеют готовые решения для проведения реакции расщепления белков: высокоспецифичные и воспроизводимые наборы; способы пробоподготовки на основе иммобилизации ферментов на магнитных частицах, а также автоматизированные процедуры. Это позволяет минимизировать ошибки ручной пробоподготовки, повысить воспроизводимость результатов и сократить время анализа.
ВЫВОДЫ. Современная методология пептидного картирования эволюционирует в сторону повышения воспроизводимости и эффективности за счет внедрения стандартизированных и автоматизированных решений для пробоподготовки при сохранении трипсина в качестве основного гидролитического агента. Информация об используемых подходах позволит исследователям ориентироваться в многообразии доступных аналитических решений, ускорить разработку методов и обеспечить надежный контроль качества белковых препаратов.
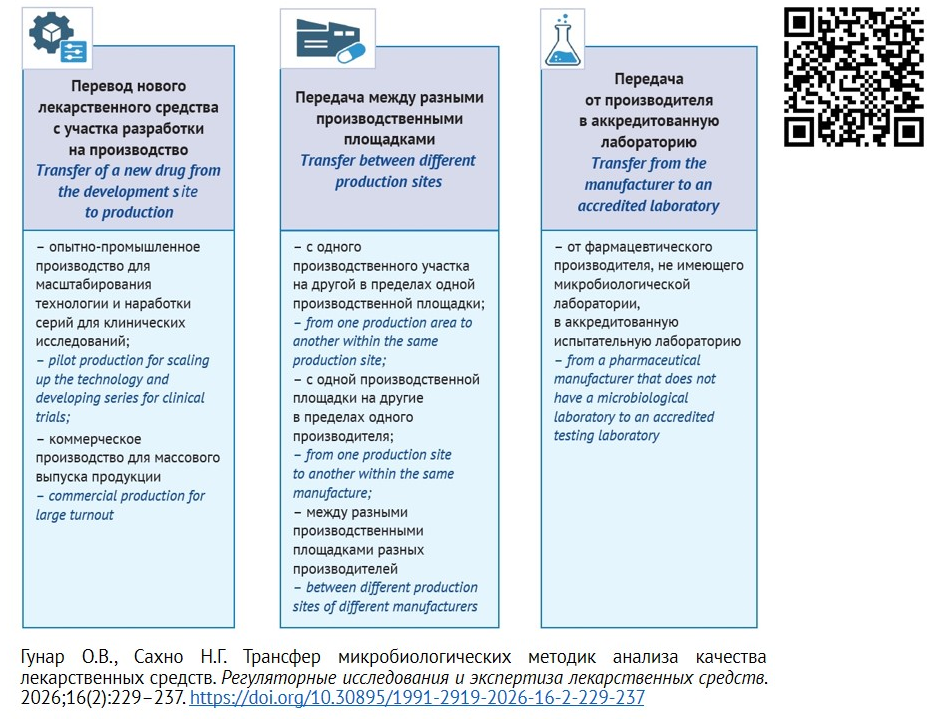
ВВЕДЕНИЕ. Трансфер микробиологических методик — важный раздел трансфера аналитических методик как части трансфера технологии между производственными площадками одной или разных фармацевтических компаний, а также в контрактные лаборатории. Регуляторные и организационные аспекты передачи аналитических методик четко регламентированы и описаны в научной литературе, в то время как трансфер микробиологических методик, не выделенный в отдельную область, часто сопряжен с рядом трудностей, например с отсутствием специализированных помещений и квалифицированного персонала, разрешения на работу с патогенными микроорганизмами и др.
ЦЕЛЬ. Создание алгоритма трансфера микробиологических методик анализа качества лекарственных средств с учетом особенностей выполнения отдельных этапов процедуры трансфера.
ОБСУЖДЕНИЕ. Трансфер методики представляет собой взаимовыгодный процесс как для принимающей лаборатории, так и для передающей стороны. При подготовке трансфера микробиологической методики стороны проводят анализ и оценку рисков для качества по ряду критериев (оснащение лаборатории, персонал, документация СМК и др.), рассматривают возможности совмещения технологий, анализируют расхождения и вносят необходимые изменения. После этого составляют план трансфера, который содержит методические особенности проведения валидационных испытаний, параметры и критерии приемлемости. Алгоритм трансфера микробиологических методик предполагает наличие аттестованных помещений, лицензии на работу с микроорганизмами 3–4 групп патогенности, обученного персонала по специальностям «Микробиология» и «Биобезопасность работы с микроорганизмами». При этом передающая и принимающая стороны могут реализовывать программы обучения персонала совместно. Фармакопейные методики в трансфере не нуждаются. При использовании альтернативной микробиологической методики требуются масштабные сравнительные валидационные исследования.
ВЫВОДЫ. Перенос микробиологических методик является наиболее критичной частью процесса трансфера на фармацевтическом предприятии, которая базируется в настоящее время на ОФС.1.1.0030 ГФ РФ, ОФС.2.3.16.0 ФЕАЭС и Руководстве ЕЭК от 08.06.2021 № 11. В процедуру трансфера могут быть внесены обоснованные дополнения, касающиеся функционирования конкретной лаборатории, включая микробиологический мониторинг, валидацию очистки и др.

ISSN 3034-3453 (Online)





























