


MAIN TOPIC: PHARMACOTHERAPY OF CARDIOVASCULAR DISEASES: DRUG DEVELOPMENT AND QUALITY CONTROL

Chronic heart failure (CHF) is one of the key sociomedical challenges that define the disease prognosis and patients’ quality of life. Modern pharmacotherapy shifts the focus from symptomatic treatment to early intervention and modified prognosis, thus necessitating a strict evidence-based/regulatory assessment and innovative drugs. In this context, an expert opinion on the evolution of CHF pharmacotherapy is of particular interest, alongside with a search for new therapeutic targets and the translation prospective of fundamental research findings into clinical practice through a lens of science, clinical experience, and regulatory reasoning. These and other questions are answered in an interview with Yury N. Belenkov, Academician of the Russian Academy of Sciences, Director of the Ostroumov Clinic for Hospital Therapy and Sechenov University Centre of Heart Failure, and President of Russian Society of Experts in Heart Failure.
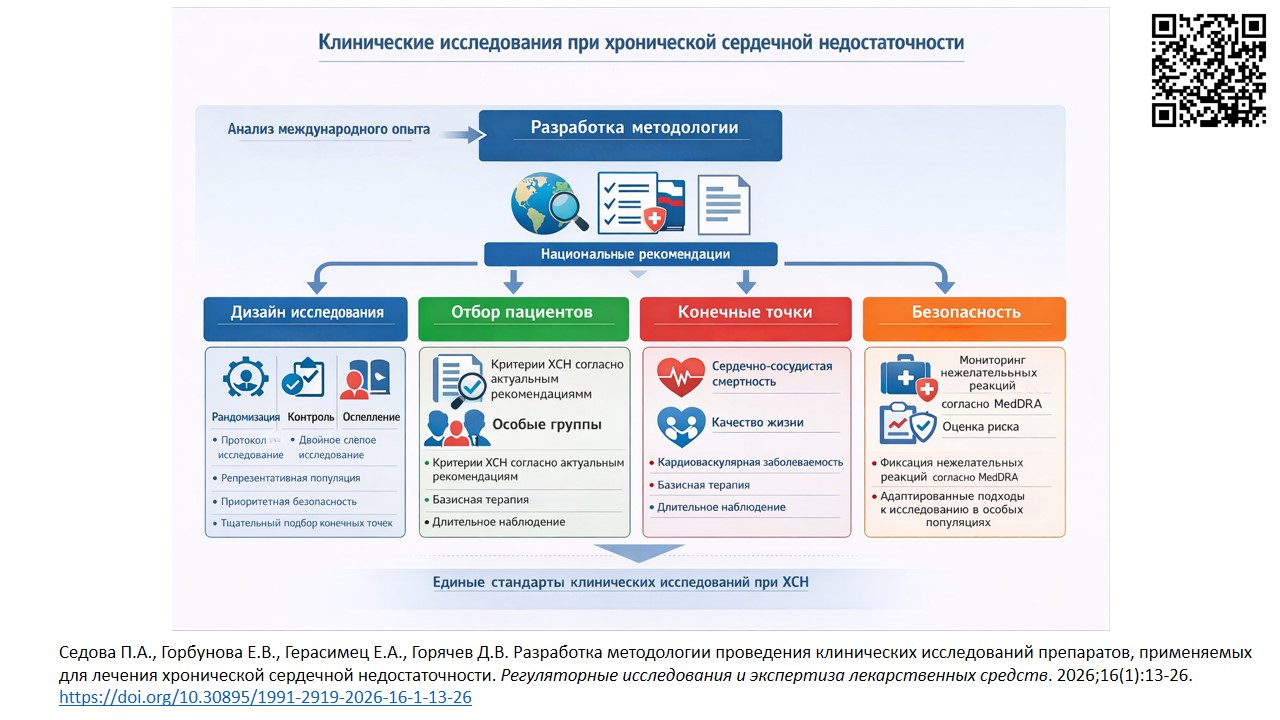
INTRODUCTION. The issue of chronic heart failure (CHF) is gaining importance not only in clinical medicine, but also for the entire health care system and society. The need to develop new drugs and drug combinations to treat CHF remains highly relevant. Currently, Russia and the EAEU countries lack regulatory documents or guidelines that control the planning, conduct and evaluation of clinical trials for CHF drugs. Therefore, it is essential to develop such a guideline considering national authorization requirements.
AIM. This study aimed to systematize international approaches to clinical trials of CHF drugs and use them as a base for the relevant guidelines effective in the Russian Federation.
DISCUSSION. Clinical trials of CHF drugs are based on such key principles as randomization, control, blinding, clinically significant endpoints, representativeness of the population, and safety prioritization. Randomized controlled trials using the double-blind method should be deemed as a measurement standard for the efficacy and safety of a new drug. To confirm therapeutic efficacy, a careful selection of patients by well-defined diagnostic criteria is warranted, alongside with a sufficient sample size and observation time, similar concomitant baseline therapy, and development of adapted approaches for special populations (elderly and pediatric patients). The endpoints are chosen by their impact on the disease prognosis and patient quality of life, as well as the contribution of the drug. Notably, increasing attention is being paid to current patient-oriented outcomes (such as improved well-being), provided that safety is ensured and there is no negative impact on the survival rate.
CONCLUSIONS. The principles of conducting CHF clinical trials both globally and in the Russian Federation are unified and stem from the fundamentals of evidence-based medicine. Implementing the described scientific principles of clinical research in CHF patients will contribute to improved treatment standards, which, in turn, should have a positive impact on the disease prognosis and outcomes.

INTRODUCTION. The approved Russian regulations and guidelines on identification of medicinal products containing Convallaria majalis cardiac glycosides use group identification tests and thin-layer chromatography; spectrophotometry and bioassay are applied to quantify glycosides. Phasing out animal tests and lack of precision and reproducibility for bioassay methods necessitate the development of new physicochemical methods for the quality control of Convallaria majalis products.
AIM. This study aimed to replace in vivo assay of cardiac glycosides in Convallaria majalis medicinal products with physicochemical analysis.
MATERIALS AND METHODS. The study objects included Russian products: Convallaria tincture and Zelenin drops; Convallaria majalis extract reference standard, convallatoxin reference standard (93.0%). Biological activity was assessed on Rana ridibunda according to a compendial method. Convallatoxin content was determined using high-performance liquid chromatography (HPLC) within our method. Spearman’s rank correlation was assessed between the results of bioassay and HPLC/HPLC and spectrophotometry.
RESULTS. The study proved validity of an HPLC method for identification and quantitation of medicinal products containing Convallaria majalis cardiac glycosides. The content of convallatoxin, the predominant and most biologically active glycoside was determined by HPLC: Convallaria majalis extract standard reference — 0.14578 mg/mL; Convallaria majalis tincture — 0.01397 mg/mL; Zelenin drops — 0.00630 mg/mL; biological activity was determined as well. Spearman’s rank correlation of the findings was performed.
CONCLUSIONS. An HPLC-based method providing precise and reproducible results has been proposed for convallatoxin identification and quantitation in Convallaria tincture and Zelenin drops. Recommended convallatoxin content: Convallaria tincture 0.01–0.02 mg/mL; Zelenin drops 0.004–0.008 mg/mL.
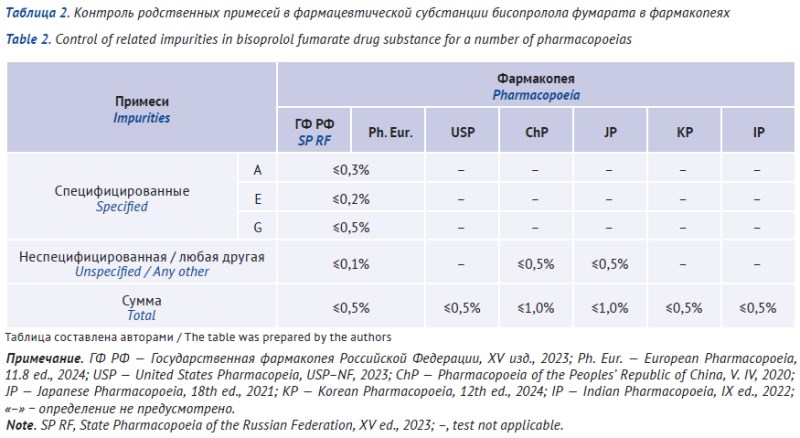
INTRODUCTION. Beta-blockers are one of the major groups of medicines used to treat cardiovascular diseases. Bisoprolol fumarate is of particular interest as it shows high selectivity towards beta-1 receptors. A monograph on bisoprolol fumarate drug substance was included in the State Pharmacopoeia of the Russian Federation, Edition XV (SP RF); however, there is no monograph on the medicinal product. Thus, it seems advisable to systematize the data on Russian and other national compendial requirements for bisoprolol fumarate medicinal product and drug substance.
AIM. This study aimed to systematize quality requirements for drug substances in order to develop the guidelines on specification drafting for bisoprolol fumarate drug substance and bisoprolol fumarate tablets.
MATERIALS AND METHODS. The study used a comparative analysis and a content analysis. The study object included the monographs of SP RF, European Pharmacopoeia (Ph. Eur.), British Pharmacopoeia (ВР), United State Pharmacopeia (USP), Pharmacopoeia of the Peoples’ Republic of China (ChP), Japanese Pharmacopoeia (JP), Korean Pharmacopoeia (KP), and Indian Pharmacopoeia (IP) for the quality of bisoprolol fumarate drug substance.
RESULTS. Monographs for bisoprolol fumarate drug substance were included in SP RF, Ph. Eur., USP, IP, ChP, JP, and KP. There was no quality specification for bisoprolol fumarate tablets in SP RF; however, the monographs for this dosage form were included in BP, USP, ChP, and JP. The monograph Bisoprolol fumarate (SP RF) was compared to other pharmacopoeias; it is generally harmonized with Ph. Eur.; however, the monographs differed with regard to authentication of the active substance and the need to control fumaric acid content. Registration dossiers were analyzed for bisoprolol fumarate drug substances and tablets. It was concluded that combinations of high-performance liquid chromatography-thin-layer chromatography or high-performance liquid chromatography-spectrophotometry can be used to assess the quality of bisoprolol fumarate and identify the active substance.
CONCLUSIONS. The performed study has justified the choice of quality parameters and analytical methods used to develop the guidelines on preparing specifications for bisoprolol fumarate tablets. The identified impurities can be described as process-related impurities resulting from the synthesis of the drug substance or degradation impurities; this is also applicable for the guidelines on specification drafting.
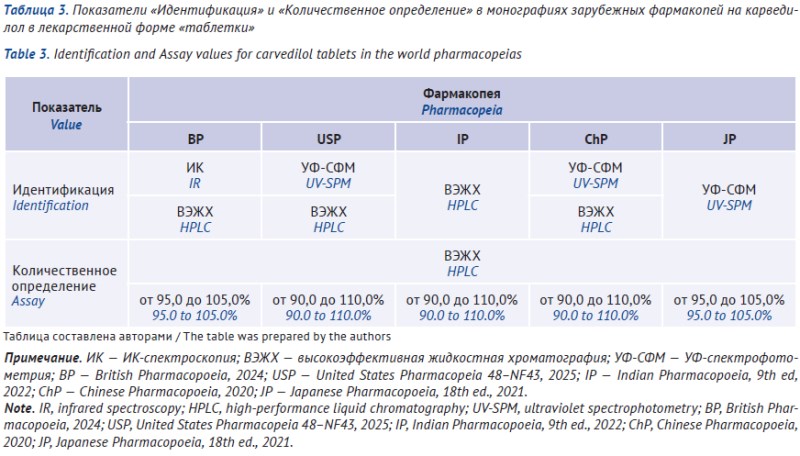
INTRODUCTION. Carvedilol is a drug used to treat renovascular hypertension and cardiovascular diseases. A monograph for carvedilol drug substance has been included in the State Pharmacopeia of the Russian Federation (SP RF); however, quality requirements lack for carvedilol products. In order to develop a relevant monograph, it appears feasible to systematize current Russian and global compendial requirements for carvedilol products.
AIM. This study aimed to develop an approach to quality control of carvedilol medicinal products (tablets) and carvedilol drug substance.
DISCUSSION. According to Russian State register of medicines, carvedilol drug substance by seven manufacturers and 18 authorized products in the form of tablets (of them, 11 authorized in the EAEU) are marketed in the Russian Federation. Carvedilol drug substance is included in SP RF, ed. XV, as well as the leading world pharmacopeias — European Pharmacopoeia (Ph. Eur.), British Pharmacopoeia (ВР), US Pharmacopeia (USP), Indian Pharmacopoeia (IP), Chinese Pharmacopoeia (ChP), Japanese Pharmacopoeia (JP), and Korean Pharmacopoeia (КР). Monographs for carvedilol tablets were included in several leading pharmacopeias — ВР, USP, IP, ChP, JP; ChP included a monograph for carvedilol capsules. The requirements were compared for key quality parameters: Identification, Related impurities, and Assay for the substance and the tablets; and Dissolution (additionally) for tablets. Standardization approaches to impurities and Dissolution test differ significantly between the pharmacopeias; basic methods to control impurities typically include high-performance liquid chromatography (HPLC); infrared spectroscopy is the basic identification method.
CONCLUSIONS. A uniform approach has been suggested based on a comparative analysis of compendial documents. The approach helps to define quality requirements for carvedilol (drug substance and tablets): choosing high-priority parameters; providing a rationale for analytical methods (infrared spectroscopy, HPLC, titrimetry/HPLC assay, and dissolution); and setting standard requirements for risk-based control of impurities. The suggested approach is applicable for expert evaluation of carvedilol dossier and monograph draft for SP RF.
DEVELOPMENT OF MEDICINES
INTRODUCTION. Oral administration of probiotics is limited by their low viability while passing through the harsh gastrointestinal environment. Microencapsulation in the alginate–chitosan system makes it possible to protect bifidobacterial cells and ensure their delivery to the large intestine. However, the effect of chitosan coating on alginate microcapsules and the relationship between the release profiles of model compounds and the viability of encapsulated probiotics remain understudied; this necessitates an experimental comparison of coating parameters, microcapsule size, and their effects on release kinetics and colony-forming unit (CFU) recovery.
AIM. This study aimed to develop guidelines for the design of probiotic dosage forms using Bifidobacterium bifidum as a case.
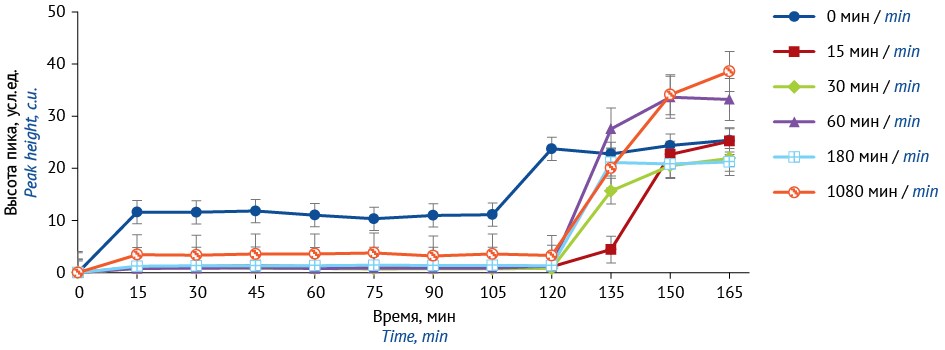
MATERIALS AND METHODS. A 2% sodium alginate solution containing either sodium metamizole (2%) or bifidobacteria (2.5×106/1.25×106 CFU/mL) was extruded into a 5% CaCl2 solution to form microcapsules subsequently coated with chitosan (0.4%, pH 6.0) by 0–1,080 min exposure to the coating solution. Capsules were produced using 0.16 and 1.8 mm needles. Dissolution test with stepwise pH change was performed without media replacement; sodium metamizole release was quantified by UV spectrophotometry (λ = 258 nm); bifidobacterial viability was assessed by counting CFU cultivated on MRS-5 agar.
RESULTS. After only 15 min of exposure, chitosan coating markedly reduced early sodium metamizole release in the dissolution test, while further coating produced no relevant additional changes. Bifidobacterial viability was as follows: 0.16 mm needles, without coating, <1%; 0.16 mm needles, with coating, ≈20%; 1.8 mm needles, with coating, ≈50%, at an initial load of 0,5×109 CFU per batch of microcapsules, and ≈80% at 1×109 CFU.
CONCLUSIONS. Short-term exposure of alginate microcapsules in chitosan forms a functional barrier that reduces premature release and increases the viability of microencapsulated probiotics; increasing capsule size further enhances the protective properties of the delivery system. Recommended baseline conditions for large-scale probiotic microencapsulation include chitosan coating with ~15 min exposure (0.4% chitosan, pH 6.0). Large extrusion needles and the final-product package with the high water and oxygen barrier (Alu-Alu blisters or dessicated bottles) is a preferred option for maximum viability and convenient dosing.

INTRODUCTION. Despite a broad range of available hypoglycemic agents, the choice of dosage forms, particularly those with prolonged release, remains limited. This necessitates the development of injectable depot formulations for the prolonged and controlled release of an active pharmaceutical ingredient. A key milestone and one of the pivotal factors in developing a stable parenteral depot formulation is testing solubility of the active pharmaceutical ingredient and selecting a solvent / solvent mixture that ensures effective release of the compound. One of the most frequently prescribed hypoglycemic agents is gliclazide, currently only available in oral dosage forms. Therefore, the development of a prolonged-release form for parenteral administration is urgent, since it will optimize therapy for a wide range of patients.
AIM. This study aimed to develop a prolonged-release depot system of gliclazide for parenteral administration.
MATERIALS AND METHODS. Gliclazide solubility in biocompatible solvents was determined according to a pharmacopeial method (under standard conditions and when heated). In vitro biopharmaceutical studies were based on dialysis with a cellophane membrane. Gliclazide in dialysate samples was quantified using UV spectrophotometry (λ=230 nm). Antidiabetic effect of the test samples was determined in male Wistar rats with an alloxan-induced diabetes model. Blood glucose was measured using an Accu-Chek® Performa Nano glucometer for 24 h after a single administration of the test samples.
RESULTS. Gliclazide solubility assessment indicated better solubility in dimethyl sulfoxide and 95% ethyl alcohol. However, gliclazide was practically insoluble in polyethylene glycol-400, propylene glycol-1,2, isopropyl myristate, glycerol, and slightly soluble in 70% ethyl alcohol. When heated, gliclazide solubility in propylene glycol-1,2 and polyethylene glycol-400 increased to slightly soluble. Gliclazide release from an aqueous suspension model over 2 h was 15.69±0.49%. Comparable antidiabetic effect was established for injection depot models using binary dispersion media based on combinations of water for injection, propylene glycol-1,2, and dimethyl sulfoxide. A more pronounced hypoglycemic effect was observed when using water for injection : propylene glycol-1,2 and dimethyl sulfoxide : propylene glycol-1,2 as a dispersion medium.
CONCLUSIONS. Model compounds have been obtained for gliclazide prolonged-release injectable depot forms based on the binary media, with a subsequent biopharmaceutical assessment. The findings necessitate the use of organic solvents in the suspension that would improve gliclazide release from the aqueous suspension.
PRECLINICAL STUDIES

INTRODUCTION. Currently, preclinical development of analgetic drugs is facing a large number of obstacles, mostly due to the limited validity of models and methods for pain assessment. To overcome the translational barrier in the development of analgetics, it is necessary to revise the existing methods for assessing pain sensitivity and develop new approaches that include both the study of reflexive and affective pain component.
AIM. This study aimed to systematize a contemporary view of pain assessment methods in laboratory animals and develop applicability criteria in preclinical trials of new analgesics.
DISCUSSION. The literature review included 75 references, among them original research and systematic reviews over the past 35 years. Pain is a multidimensional phenomenon that includes sensory, discriminatory and affective-motivational components. Standard nociceptive tests effectively evaluate sensory hypersensitivity, however, they are not sensitive enough when studying chronic pain based on an affective component. Less widely used non-reflexive methods (grimace scale, ultrasound vocalization, burrowing test) allow assessing the affective component; still, they have low specificity and are insufficiently validated for various pain models. A combined, polymodal approach enhances the objectivity, reproducibility, and translational predictivity of preclinical research towards the development of new analgetics.
CONCLUSIONS. Nociceptive tests are a tool for assessing efficacy of anesthetics both at a primary screening and in preclinical trials. Standard nociceptive tests do not allow assessing affective pain component, thus development of new anesthetics necessitates an introduction of non-reflexive pain assessment in the preclinical trials. A combination of reflexive and non-reflexive pain assessment methods is a base for new research strategies in preclinical trials.
QUALITY CONTROL OF MEDICINES
INTRODUCTION. Amendments to Federal Law No. 61-FZ On Circulation of Medicines provide for additional data on excipients in general pharmacopeial monographs and pharmacopeial monographs; this necessitates the improvement of standardization requirements for excipients at a legislative level.
AIM. This study aimed to determine the main growth vectors of pharmacopeial approaches to excipient standardization, as exemplified by propylene glycol and its derivatives.
DISCUSSION. The study considered the concept of excipients and their standardization requirements at the national, regional, and international levels. It was established that the term “substances for pharmaceutical use” most correctly describes pharmacopeial approaches to excipient standardization. It was revealed that the number of excipient names for medicinal products available on the Russian pharmaceutical market remains a relevant issue. Approaches from the State Pharmacopoeia of the Russian Federation, the Pharmacopoeia of the Eurasian Economic Union, and foreign pharmacopeias (US, Europe, China) were compared regarding standardization of general quality requirements for excipients and specific requirements for propylene glycol and its derivatives. It was established that pharmacopeial requirements for excipients are based on an integrated approach that defines general quality parameters for the substances and assesses additional parameters that depend on the performance. A list of performance parameters was compared for various national pharmacopeias; noteworthy, the designations on the list were not always standardized.
CONCLUSIONS. The pharmacopeial approach to excipient standardization is a critical tool for ensuring quality, efficiency, and safety of medicinal products. Based on the analysis of quality control data for propylene glycol and its derivatives, key pharmacopeial methods of excipient standardization include: developing and updating monographs for excipients; improving the requirements for universal quality parameters of excipients as per monograph Substances for Pharmaceutical Use and establishing performance requirements; expanding the range of excipients included in the State Pharmacopoeia of the Russian Federation; and increasing the number of pharmacopeial standards for certain excipients upon analysis and harmonization of national, regional, and international requirements.
INTRODUCTION. Lack of national requirements regulating the quality of valacyclovir hydrochloride active pharmaceutical ingredient (API) in hydrated form and valacyclovir medicinal products may be challenging not only for manufacturers, but also for experts examining submitted materials in registration dossiers. In this regard, it seems relevant to analyze the pharmacopoeial requirements for the quality of the pharmaceutical substance valacyclovir hydrochloride and medicinal products containing valacyclovir for applicants preparing the registration dossier and for experts examining the quality of medicinal products.
AIM. This study aimed to analyze and generalize quality requirements for valacyclovir medicinal products: pharmaceutical substance in anhydrous and hydrated form and tablets.
MATERIALS AND METHODS. The study compared quality requirements for valacyclovir hydrochloride API described in the State Pharmacopoeia of the Russian Federation, Edition XV (SP RF), European (Ph. Eur.), British (ВР), US (USP), Indian (IP), Chinese (ChP), and Japanese Pharmacopoeias (JP), as well as registration dossiers of valacyclovir hydrochloride tablets described in BP, USP, IP, ChP, and JP monographs.
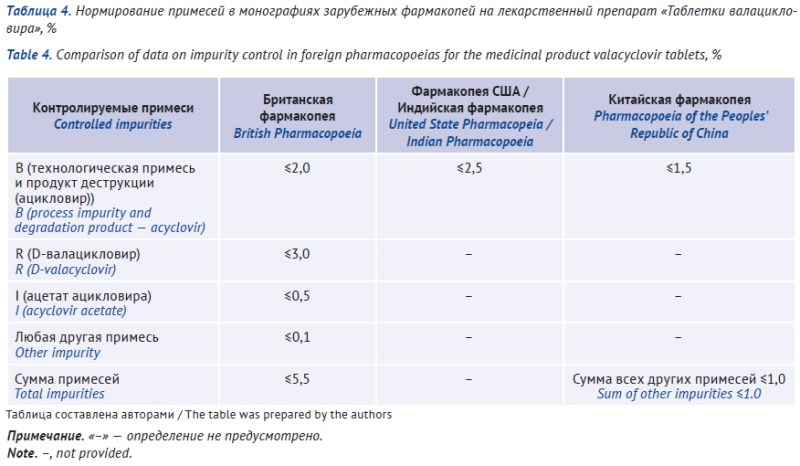
RESULTS. Valacyclovir hydrochloride is a medicinal product used to treat herpesvirus infections. Russian State Register of Medicines includes APIs from 6 manufacturers and 17 products in the dosage form of film-coated tablets (of them 14 registered in the Eurasian Economic Union). A pharmacopoeial monograph on valacyclovir hydrochloride API is included in the SP RF, Ph. Eur., BP, USP, IP, ChP, and JP. Ph. Eur., BP, USP, and IP establish requirements for both anhydrous and hydrated forms of valacyclovir. In Ph. Eur. (BP), the impurity profile of anhydrous and hydrated forms of valacyclovir differs significantly, while other pharmacopoeias have different standards of water content. Valacyclovir tablets are described in BP, USP, IP, ChP, and JP. For this dosage form, either IR spectrometry method or both HPLC and UV spectrophotometry methods should be used for identification; it is recommended to specify the identified acyclovir impurity, unspecified impurities and a sum of impurities (organic impurities). Despite varying impurity profiles for hydrated API and anhydrous tablets, BP requirements are identical, no matter what substance is used.
CONCLUSIONS. Comparative analysis of national pharmacopoeias and registration dossiers allows us to summarize the requirements for impurity control in valacyclovir API and tablets with different synthesis methods. Alternative methods have been found for identifying the API in valacyclovir products. A comparative analysis reveals differences in dissolution test standards across pharmacopoeias. Manufacturers can apply this point in the specifications, since this test confirms stability of the manufacturing process, batch homogeneity, and drug quality, rather than its comparability with comparator drugs.

ISSN 3034-3453 (Online)





























