


MAIN TOPIC: CIVIL COMMERCE REGULATION IN THE EAEU: MARKETING AUTHORISATION AND QUALITY CONTROL

31 December 2025 is the deadline for proceeding to the rules of civil commerce accepted within the Eurasian Economic Union (EAEU). According to Ekaterina M. Rychikhina, Head of Department for evaluation management and control, Scientific Centre for Expert Evaluation of Medicinal Products, the end of the transitory period is not the finishing line, but rather a new stage of development. In her exclusive interview, the expert with an unrivalled practical experience highlights the hidden risks that can emerge even if the dossier was successfully aligned, and emphasizes critical aspects that warrant particular attention from the regulatory bodies and the manufacturers. Special attention is placed on the latest amendments to the pharmaceutical civil commerce rules in the EAEU that already shape the new legal reality. The interview both announces the official position of the Centre and gives practical considerations that will help manufacturing companies avoid the mistakes and effectively interact with an expert body.
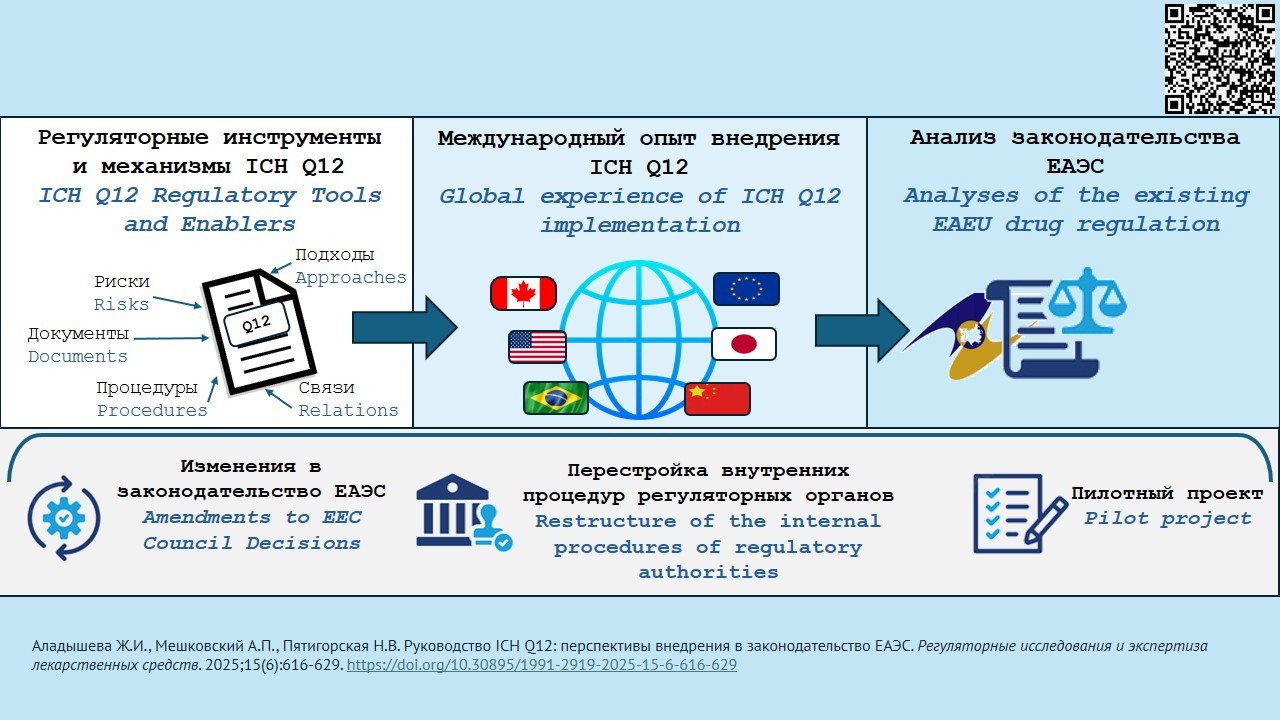
INTRODUCTION. In 2019, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) adopted the new ICH Q12 guideline on pharmaceutical product lifecycle management. It covers all change categories listed in Module 3 (Quality) of drug Registration dossiers and Master files for the pharmaceutical substance and other products. Its application is expected to help optimise change management, reduce approval and implementation time, and mitigate the risks for patients. As of August 1, 2025, the document is considered implemented in three ICH member countries: USA, Japan, and China.
AIM. This study aimed to discuss implementation prospects of ICH Q12 guideline in the EAEU legislation.
DISCUSSION. Eight tools and mechanisms recommended for post-approval change management regarding quality were assessed for their novelty in relation to existing commonly accepted principles and approaches to drug quality regulation. The analysis included experience of ICH member countries that have implemented or are implementing the guidance; pilot projects conducted; and changes related to legislation and internal procedures. So far, only the USA managed a full-fledged implementation of the document. The authors have considered the experience of pharmaceutical companies that made changes according to ICH Q12. Analysed current EAEU legislation regarding regulation of pharmaceutical products has shown that, in order to effectively apply the regulatory tools and mechanisms of the ICH Q12 Guideline, a number of fundamental amendments to Eurasian Economic Council documents (Decisions No. 77, 78, 84, and 91) is warranted, as well as an EAEU-adapted version of the Q12 Guideline, the restructured internal procedures of regulatory authorities and their subordinate expert institutions, and a pilot project to test the new administrative procedures.
CONCLUSIONS. The Q12 guidance will help transform quality change management for approved pharmaceuticals, currently a bureaucratic hurdle, into a mechanism for innovation, scientific and technological progress, and continuous product improvement. Implementing the guidance will require both legislative modifications and improved internal procedures of regulatory bodies and pharmaceutical companies. Among the proposed Q12 mechanisms, change management protocols have been the most successful experience. The Eurasian Economic Union (EAEU) provides all the basic prerequisites for Q12 implementation; however, a number of fundamental and technical issues remains unresolved, including national drug regulation systems of the EAEU member states, changes to existing documents, and the development of new ones.
DIGITAL TECHNOLOGIES AND PHARMACY

INTRODUCTION. Current methods of handling medicine regulatory documents are associated with high time cost (40-60% of labour hours), frequent documentation errors, and limited data interoperability. Neural network technologies have enabled the enhanced document preparation and a transition to full automation of the registration dossier life cycle.
AIM. This study aimed to evaluate the possibility of using artificial intelligence (AI) systems in preparing a drug registration dossier.
DISCUSSION. Natural language processing (NLP) models demonstrate high efficiency for the regulatory documentation. Named entity recognition (NER) systems with 89–96% entity extraction accuracy rate reduces the processing (preparation and quality review) time for documents within electronic Common Technical Document (eCTD) by 64%, but face limitations in interpreting morphologically complex terms and require annotated datasets. Without additional fine-tuning, generative models such as GPT-4, are prone to generating inaccurate facts when used in the Retrieval-Augmented Generation (RAG) architecture. Predictive systems based on graph neural networks and XGBoost ensembles demonstrate high accuracy (ROC AUC up to 0.88) when predicting drug approval; however, they cannot interpret decisions and data systematic biases. Developing document-centric platforms with NLP reduces the dossier preparation time by 60%, still, implementing an automated procedure for generating dossier sections requires an expert verification.
CONCLUSIONS. The concept of integrated AI systems proves its effectiveness by reducing the document handling time by manufacturers and increasing the accuracy of decisions, which in turn speeds up the market launch of medicinal products. The prospects of introducing digital technologies are associated with overcoming definitions differences through unified ontologies. Practical implementation requires the development of unified standards for the validation of AI algorithms and adaptive systems.
DEVELOPMENT OF MEDICINES
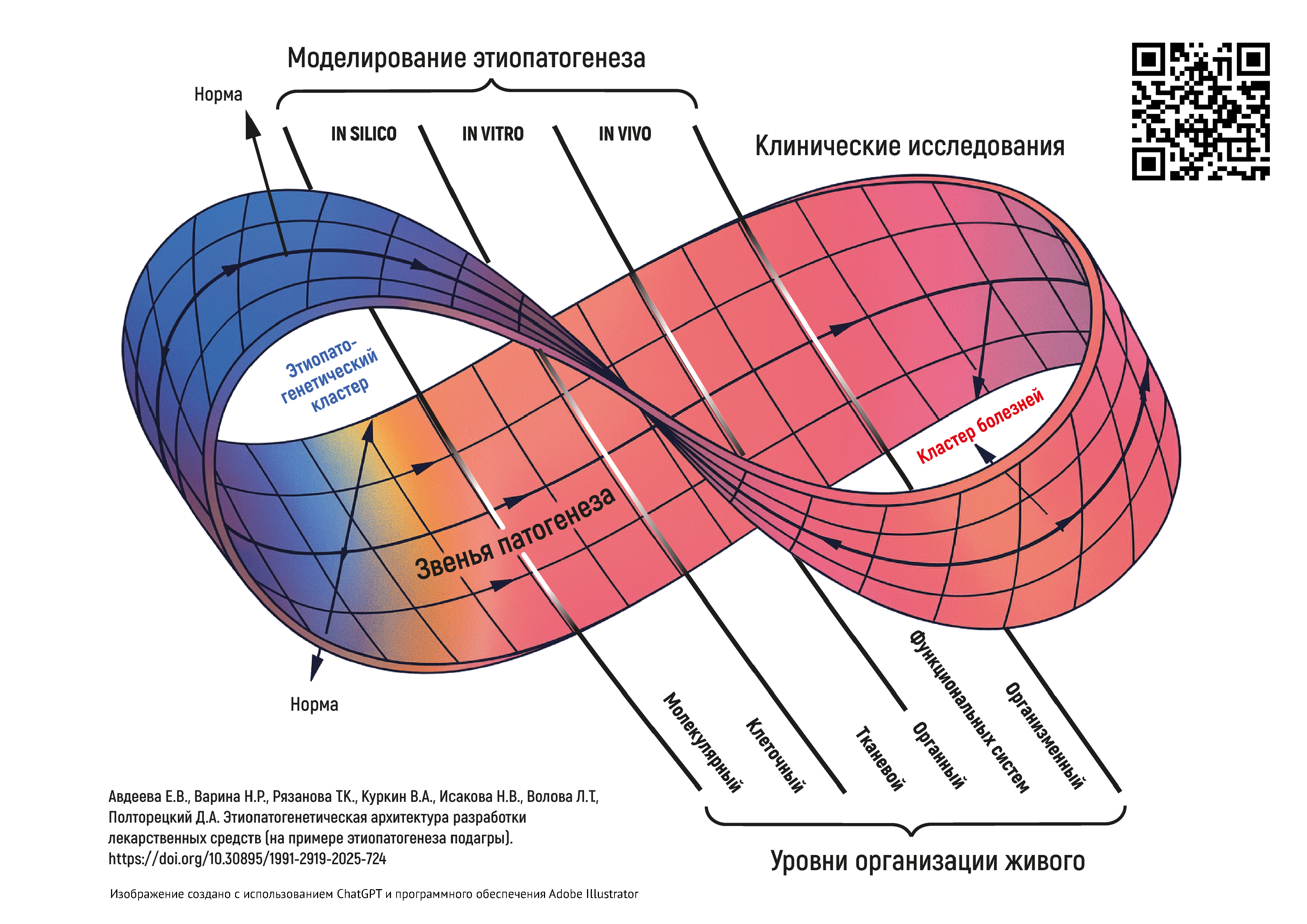
INTRODUCTION. A holistic understanding of the physiological and biochemical pathways involved in pathogenesis is needed both for doctors diagnosing and treating patients and for drug developers. The accumulated knowledge in medicine and related fields, combined with the rapid development of digital tools, enables simulating the response systems of the body under normal and pathological conditions at a qualitatively new level. Being able to perform such simulations will lead to creating a digital architecture of body conditions, with interconnected links in the chain of pathogenesis being the focal points for researchers advancing medicines from early development to clinical trials.
AIM. This study aimed to review the existing approaches that could form a foundation for constructing an aetiopathogenetic architecture of pathological conditions and diseases that would serve as a framework for targeted drug development.
DISCUSSION. Using gout as a case study, the authors demonstrated the necessity and possibility of developing a three-dimensional aetiopathogenetic architecture of pathological conditions and diseases that would be based on the hierarchical relationships of pathological processes at different biological organisation levels. The study identified key applications for the aetiopathogenetic architecture. In medicine, the aetiopathogenetic architecture could be used in data-driven individual diagnostics and personalised pharmacotherapy. In pharmaceutics, the aetiopathogenetic architecture could provide a platform for investigating pharmacodynamics, from screening candidate compounds to applying targeted and multitargeted approaches in pharmaceutical development. The authors used the aetiopathogenetic architecture of gout as a case study to discuss the logic behind designing studies of medicines.
CONCLUSIONS. The article proposes a methodology for constructing an aetiopathogenetic architecture reflecting cause-and-effect relationships of different significance to the development of pathological conditions and diseases. The aetiopathogenetic approach should become an integrative framework for all stages of the development and use of novel medicines, as well as a basis for expanding the indications for existing medicines. New opportunities are arising for the development of aetiopathogenetic models of varying complexity that can be used in projects ranging from drug design at the molecular level to pathophysiological modelling at the organismal level.
QUALITY CONTROL
INTRODUCTION. An effective quality control system for medicinal products is impossible without reference standards used for validation and verification of analytical procedures. It is especially relevant to develop reference standards for quality control of new active pharmaceutical ingredients used to develop medicinal products.
AIM. This study aimed to develop a method for obtaining and certifying a primary reference standard of 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F] xanthine to be used in quality control of medicinal products.
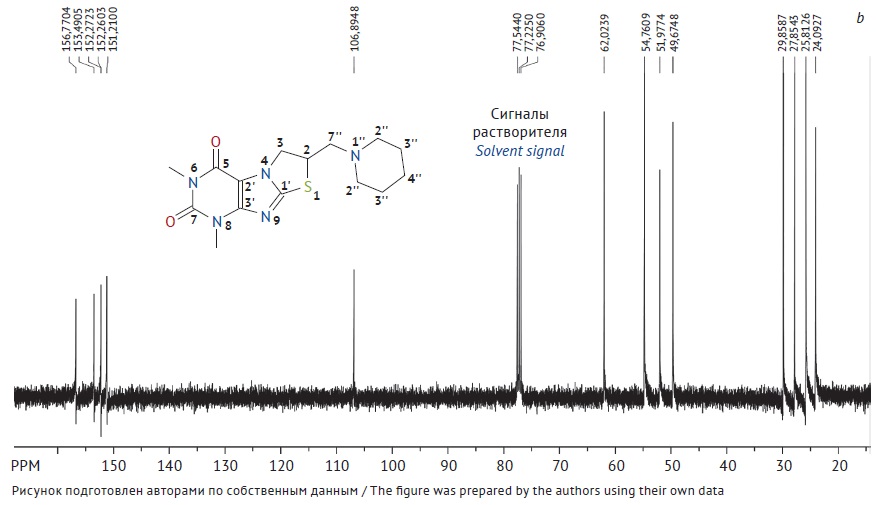
MATERIALS AND METHODS. A reference standard was obtained by recrystallising 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine from ethanol. The structure of the reference standard was defined by nuclear magnetic resonance and infrared spectroscopy; purity was measured by mass balance, high-performance liquid chromatography (related impurities), and titration (non-aqueous acidimetry).
RESULTS. The study assessed physical and chemical properties of a reference standard for 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine. This included structure elucidation by infrared and nuclear magnetic resonance spectroscopy; loss on drying 0.073±0.015%; sulfate ash 0.080±0.009%; related substances (not found); heavy metal impurities (not more than 0.002%); elemental composition (C — 53.68±0.17%; H — 6.32±0.02%; N — 20.81±0.09%; O — 9.52±0.06%; S — 9.57±0.04%); quantitative determination by non-aqueous titration 99.74±0.12%, and mass balance 99.85±0.01%.
INTRODUCTION. An effective quality control system for medicinal products is impossible without reference standards used for validation and verification of analytical procedures. It is especially relevant to develop reference standards for quality control of new active pharmaceutical ingredients used to develop medicinal products.
AIM. This study aimed to develop a method for obtaining and certifying a primary reference standard of 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F] xanthine to be used in quality control of medicinal products.
MATERIALS AND METHODS. A reference standard was obtained by recrystallising 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine from ethanol. The structure of the reference standard was defined by nuclear magnetic resonance and infrared spectroscopy; purity was measured by mass balance, high-performance liquid chromatography (related impurities), and titration (non-aqueous acidimetry).
RESULTS. The study assessed physical and chemical properties of a reference standard for 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine. This included structure elucidation by infrared and nuclear magnetic resonance spectroscopy; loss on drying 0.073±0.015%; sulfate ash 0.080±0.009%; related substances (not found); heavy metal impurities (not more than 0.002%); elemental composition (C — 53.68±0.17%; H — 6.32±0.02%; N — 20.81±0.09%; O — 9.52±0.06%; S — 9.57±0.04%); quantitative determination by non-aqueous titration 99.74±0.12%, and mass balance 99.85±0.01%.
INTRODUCTION. An effective quality control system for medicinal products is impossible without reference standards used for validation and verification of analytical procedures. It is especially relevant to develop reference standards for quality control of new active pharmaceutical ingredients used to develop medicinal products.
AIM. This study aimed to develop a method for obtaining and certifying a primary reference standard of 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F] xanthine to be used in quality control of medicinal products.
MATERIALS AND METHODS. A reference standard was obtained by recrystallising 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine from ethanol. The structure of the reference standard was defined by nuclear magnetic resonance and infrared spectroscopy; purity was measured by mass balance, high-performance liquid chromatography (related impurities), and titration (non-aqueous acidimetry).
RESULTS. The study assessed physical and chemical properties of a reference standard for 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine. This included structure elucidation by infrared and nuclear magnetic resonance spectroscopy; loss on drying 0.073±0.015%; sulfate ash 0.080±0.009%; related substances (not found); heavy metal impurities (not more than 0.002%); elemental composition (C — 53.68±0.17%; H — 6.32±0.02%; N — 20.81±0.09%; O — 9.52±0.06%; S — 9.57±0.04%); quantitative determination by non-aqueous titration 99.74±0.12%, and mass balance 99.85±0.01%.
CONCLUSION. We have developed production process of 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine reference standard. Physical and chemical properties of the above standard comply with the requirements and allow us to recommend 6,8-dimethyl-2-piperidinomethyl-2,3-dihydrothiazolo[2,3-F]xanthine as a reference in the quality control of medicinal products.
INTRODUCTION. low-toxic polyphenolic compound with significant antioxidant activity. Genistein Genistein (5,7-dihydroxy-3-(4-hydroxyphenyl)chromene-4-on) is in combination with chemoradiotherapy can prevent and reduce radiation damage. However, in order to be included in the prevention and treatment regimens of radiation injuries, genistein should be authorised as a medicinal product. This warrants assessment methods of genistein content that would meet pharmacopoeial requirements for analytical methods.
AIM. This study aimed to develop a method of high performance liquid chromatography (HPLC) for identification and quantitation of the potential active pharmaceutical substance genistein.
MATERIALS AND METHODS. Samples of active pharmaceutical substance genistein were synthesised in St. Petersburg State Chemical and Pharmaceutical University. All samples were analysed using Prominence LC-20 Shimadzu HPLC system with diode array detection (Japan). The temperature in the column compartment was 40 °C, the injection volume 100 μL, the detection wavelength 261 nm. A Shimadzu C18 column (4.6 mm×250 cm, 5 μm particle size) was selected for analysis.
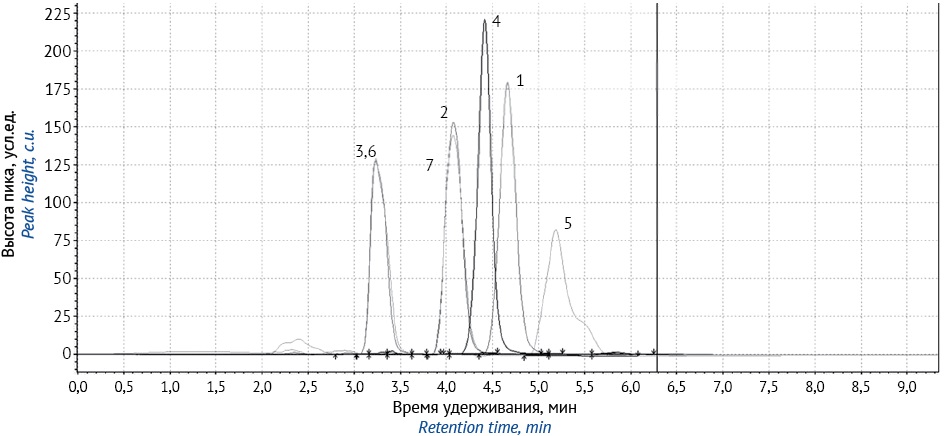
RESULTS. A method was developed for identification and quantitation of a potentially active pharmaceutical substance by HPLC with diode array detection using a mixture of acetonitrile and purified water (75:25 v.:v.) as a mobile phase. The flow rate was 0.7 mL/min and the column temperature 40 °C. Applicability of the developed system was evaluated. During optimisation of the conditions, the flow rate of the mobile phase was set to 0.7 mL/min. The suitability of the developed system was evaluated. The method was validated according to the following parameters: specificity, linearity (0.001–0.01%; r=0.997), correctness (RSD%=1.26), reproducibility (RSD%=0.43), intraday precision (RSD%<2), and robustness.
CONCLUSIONS. The developed method of genistein identification and quantitation by HPLC with diode array detection meets pharmacopoeial requirements for analytical methods and can be proposed for inclusion in the draft pharmacopoeial monograph “Genistein”.

INTRODUCTION. Beta-glucans and peptidoglycans are cell wall components of bacteria and fungi that are potential sources of pyrogenic contamination of parenteral medicines. Such impurities can cause adverse immune reactions. Therefore, recently, certain attention has been paid to identification of beta-glucans and peptidoglycans in medicines. Despite the lack of a harmonised detection method for beta-glucans and/or peptidoglycans, pharmaceutical industry uses several methods for identification and quantitation of these impurities.
AIM. This study aimed to assess applicability of the existing detection methods for beta-glucans and / or peptidglycans in the medicinal products.
MATERIALS AND METHODS. Applicability of detection was examined for beta-glucans and peptidoglycans using amoebocyte lysate and silkworm larvae plasma as reagents. Beta-glucans were detected in Bupivacaine, the product that was previously found to have random glucan impurities. In the drug tests, three types of amoebocyte lysate reagents of different compositions were used reacting to 1) bacterial endotoxins and beta-glucans (lysate with factors C and G); 2) only bacterial endotoxins (lysate with factor C); 3) only beta-glucans (lysate with factor G). Peptidoglycans in Icodextrin were assessed using a reagent from the silkworm larvae plasma. For qualitative analysis, colour of the test solutions was visually assessed after heating them in a dry-air block heater. Kinetic photocolorimetric method was used for quantitation; the primary data were processed using R software.
RESULTS. As a result of two tests with amoebocyte lysate reagents (factors C and G) and (factor C), beta-glucans were detected in Bupivacaine. Chromogenic kinetic method using amoebocyte lysate (factor G) quantified the impurity, which exceeded 2,000 pg/mL. For Icodextrin, peptidoglycan reference content (not more than 200 pg/mL) was not exceeded.
CONCLUSIONS. Identification methods for beta-glucans and/or peptidoglycans using amoebocyte lysate and silkworm plasma are applicable for identifying these impurities in the medicinal products. While choosing a study method, product composition and analytical purpose are to be considered.
HERBAL MEDICINAL PRODUCTS

INTRODUCTION. Herbal extraction products are commonly used in medical practice. Elemental impurities is one of their key quality indicators. Process technology is among the factors influencing the final product’s composition. Transfer specifics of elements, including heavy metals and As, from the initial plant material into extraction-based dosage are poorly highlighted.
AIM. This study aimed to assess the extraction of elemental impurities from medicinal plant materials into tinctures extracted by different methods.
MATERIALS AND METHODS. The study object was motherwort herb used for the industrial production of tinctures (manufacturer — OAO Flora Kavkaza). Tinctures were prepared under laboratory conditions using fractional maceration, ultrasound-assisted extraction, and vortex extraction. The elemental composition of the initial plant material and the obtained tinctures were analysed by inductively coupled plasma mass spectrometry (ICP-MS) using Agilent ICP-MS 7900.
RESULTS. Thirteen elements (V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Sr, Cd, Tl, Pb) were identified in the motherwort herb and its tinctures; Hg was not detected. Elemental concentrations in the initial raw material ranged from 0.007 to 121.098 mg/kg, whereas the toxic elements (Pb, Cd, As) complied with compendial requirements. In the tinctures, the heavy metals did not exceed 1.25 mg/kg; Zn, Cu, and Mn were present in higher concentrations, while Tl and Cd were found in minimal amounts. Potential intake of elements within the studied extracts was assessed, as well as their medical safety. The transfer rates of heavy metals and arsenic from the raw material into the obtained tinctures were calculated; for most elements, they did not exceed 46%. It was established that Zn was extracted into all test tinctures in the highest quantities, while Cd was transferred in the smallest amounts. When using fractional maceration, Zn, Ni, Cu, and Tl entered the tinctures in the highest quantities; vortex extraction resulted in the highest transfer of V, Cr, Co, and Sr. Ultrasound-assisted extraction resulted in the lowest amount of heavy metal impurities entering the solution.
CONCLUSIONS. The patterns of elemental transfer into hydroalcoholic extracts with different extraction methods were studied using motherwort tinctures as an example. It was established that fractional maceration and vortex extraction yields tinctures with a higher content of essential elements, whereas the use of ultrasound-assisted extraction results in minimum concentrations of toxic heavy metal impurities (Pb, Cd) and As.
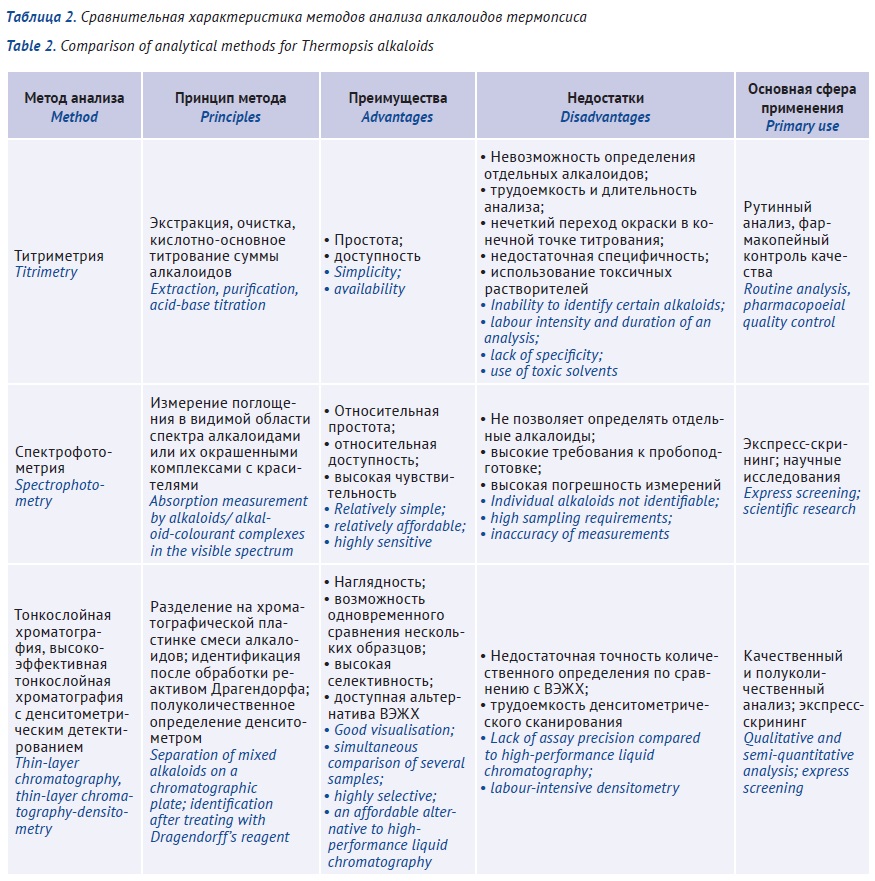
INTRODUCTION. The major alkaloids of Thermopsis lanceolata R. Br. herb are thermopsine (N-methylcytisine), pachycarpine, and anagyrine, while the seeds predominantly contain cytisine used to treat tobacco addiction. Differing toxicity and pharmacological activity of separate alkaloids requires both control of their total content and monitoring of each individual alkaloid.
AIM. This study aimed to prospectively assess the use of certain analytical methods for quantitation of Thermopsis alkaloids.
DISCUSSION. Pharmacopoeial titrimetric method is labour-intensive, time-consuming, has low detection accuracy of the test solution at the titration endpoint, uses toxic solvents, and, most importantly, only determines the sum of alkaloids without differentiating them. Spectrophotometric methods as well do not allow for the reliable determination of individual alkaloids due to the lack of selectivity (spectral overlap for certain alkaloids), and the determination depends heavily on the sampling conditions. So far, the most reliable instrumental methods for Thermopsis herb assay are thin-layer chromatography (TLC) and high-performance liquid chromatography (HPLC).
CONCLUSIONS. TLC and HPLC methods can be used in pharmacopoeial analysis to isolate and quantify individual alkaloids (identify cytisine and thermopsine), as well as to identify the impurities from Thermopsis seeds in medicinal products not intended for the treatment of tobacco addiction.
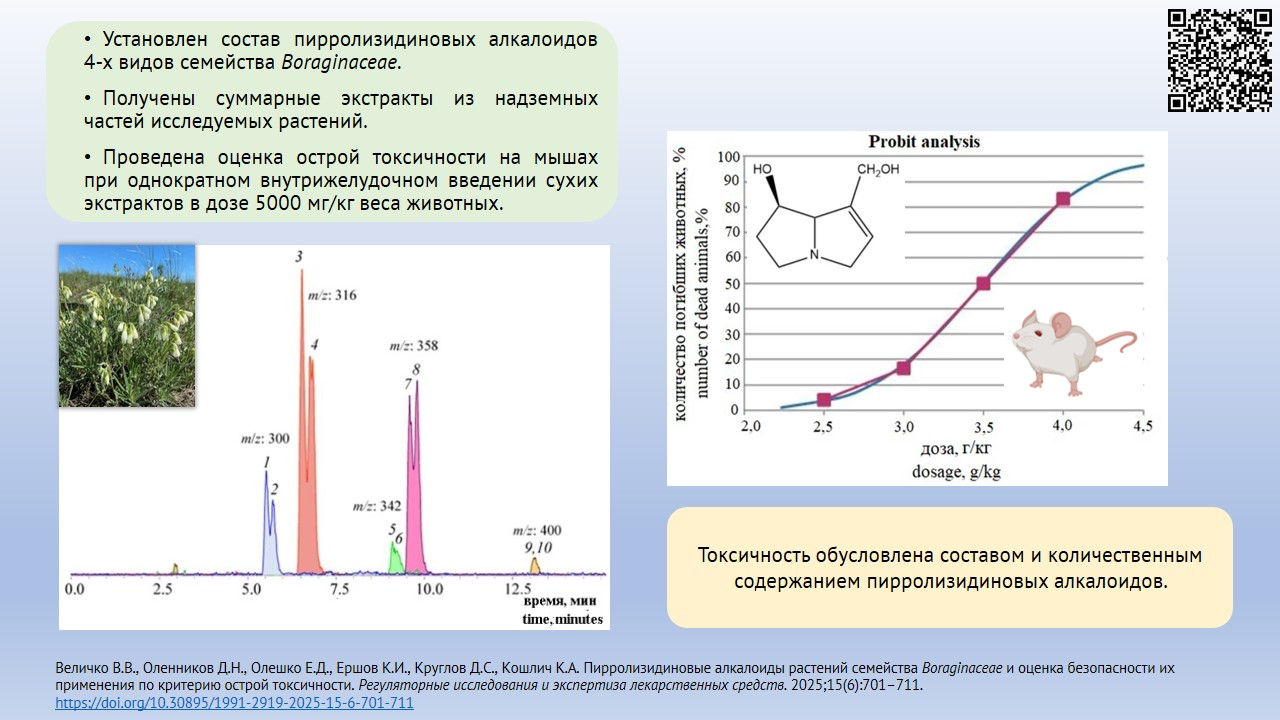
INTRODUCTION. Plants of Boraginaceae family (Pulmonaria mollis, Nonea rossica, Onosma simplicissima, and Cynoglossum officinale) are widespread over the Russian Federation and are promising sources of phytomedicines with important pharmacological properties, such as antimicrobial, antianaemic, anticoagulant etc. Currently, Boraginaceae are not classified as officinal plants, presumably due to pyrrolizidine alkaloids (PA) that can cause hepatotoxic effect.
AIM. This study aimed to assess safety using Boraginaceae plants based on acute toxicity and content of pyrrolizidine alkaloids.
MATERIALS AND METHODS. The research objects were dried herbs of Pulmonaria mollis, Nonea rossica, Onosma simplicissima, and Cynoglossum officinale, collected from flowering plants in Novosibirsk region over 2023–2024. The composition and amount of alkaloids in alcohol extracts were determined by high-performance liquid chromatography with diode array and mass-spectrometry detection with electrospray ionisation. Acute toxicity was tested in vivo in 102 mature male and female CD-1 mice weighing 24.0±2.0 g, aged 12 weeks, taken from Conventional Animal Vivarium of Scientific Center of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences. The animals received a single dose of dried study extracts diluted in distilled water at 5 g/kg.
RESULTS. The absence of PA in P. mollis herb was established, alongside with its trace amounts (0.01 μg/g) in P. mollis leaves. In other studied plant species, PA, such as enantiomers of intermedine, lycopsamine and their derivatives were found: O. simplicissima herb — 1.07±0.03 μg/g, N. rossica herb — 8.25±0.08 μg/g; in C. officinale herb — 676.3±7.4 μg/g. Assessed acute toxicity made it possible to classify dry extracts from P. mollis herbs and leaves, N. rossica herb and O. simplicissima herb as toxicity class 5, and C. officinale herb as toxicity class 4.
CONCLUSIONS. Study doses of extracts taken from herbs and leaves of P. mollis are non-toxic. For extracts from O. simplicissima and N. rossica herb, further research is relevant to determine toxicity in prolonged use. Extracts from C. officinale herb are toxic and cannot be used per os.
PHARMACY COMPOUNDING
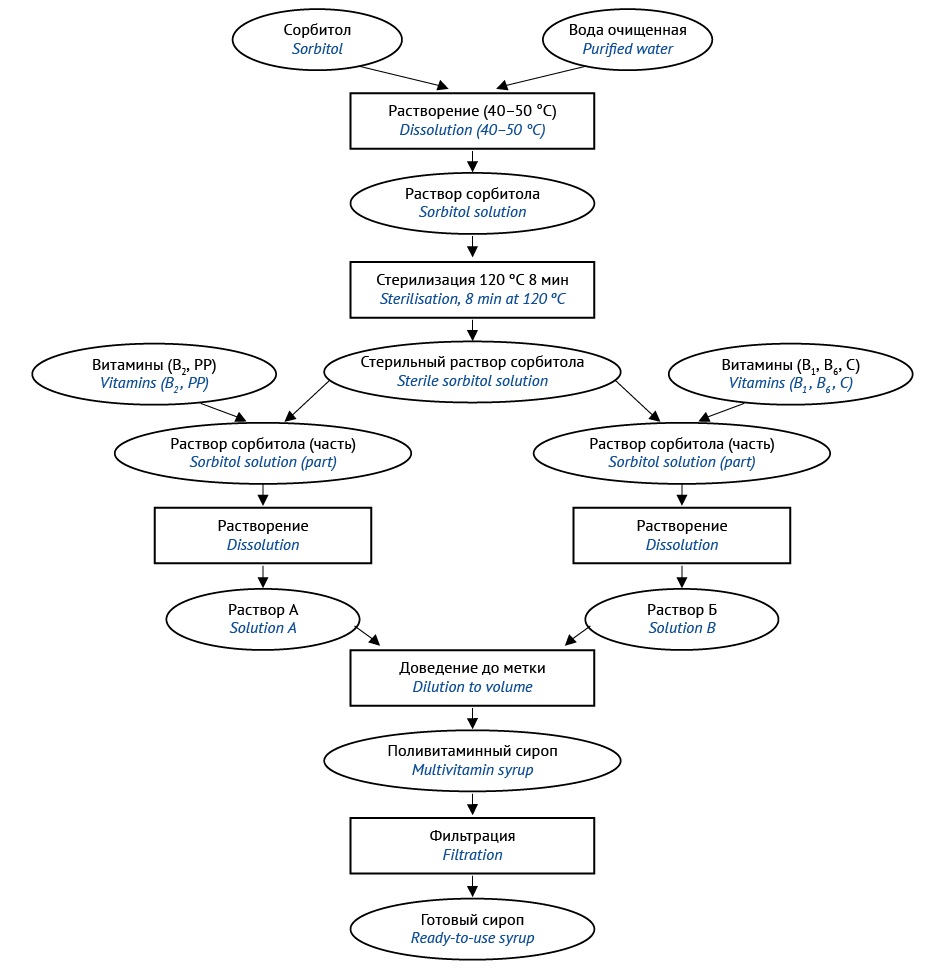
INTRODUCTION. Modern pharmaceutical market mainly offers the range of multivita min liquid medicinal products for children aged 1 to 3 years as dietary supplements. They contain excipients (preservatives, sucrose, synthetic flavours and dyes) unsafe for use in young children. Extemporaneous medicinal products allow minimising the amount of excipients undesirable for children. Some promising ways to improve small-scale extemporaneous preparation include unified prescriptions, modified process, and reasonably extended shelf life.
AIM. This study aimed to optimise formulation and production process of an extemporaneous multivitamin for paediatric patients.
MATERIALS AND METHODS. The research object was sorbitol-based multivitamin syrup prepared extemporaneously that contains ascorbic acid, nicotinic acid, thiamine hydrochloride, riboflavin, pyridoxine hydrochloride (vitamins С, РР, В1, В2, В6) in the concentrations meeting physiological needs of young children aged 1 to 3 years. The authors used active pharmaceutical ingredients that meet regulatory requirements for the above vitamins and sorbitol. Components were identified by specific quality tests; assay of the components included chemical and physico-chemical methods (spectrophotometry, spectrofluorimetry, and refractometry), adapted considering their total content. Both process parameters of the syrup (density, pH values) and organoleptic parameters (taste, smell, and colour) were part of the analysis.
RESULTS. Two syrup production methods have been developed for industrial pharmacies: technology 1 — based on a sterile sorbitol solution, technology 2 — steri lising the finished product with membrane filtration. For both products, identification and assay of all ingredients was carried out. Neither process parameters (density, pH) nor organoleptic properties (colour, smell, taste) changed within 30 days of storage at a cool place without direct sunlight.
CONCLUSIONS. Two process options have been proposed for sorbitol-based syrup containing water-soluble vitamins C, PP, B1, B2, B6 at concentrations suitable for physiological needs of children aged 1 to 3 years. Both technologies can be used in industrial pharmacies. However, using membrane filtration increases the production net cost, since it uses laboratory equipment in the pharmacy (a vacuum filtration system and a vacuum pump).

ISSN 3034-3453 (Online)





























