


MAIN TOPIC: MEDICAL TECHNOLOGY TRANSFER: INSTRUMENTS AND MECHANISMS FOR ACHIEVING PHARMACEUTICAL SOVEREIGNTY

The key mission of medical science is the development of advanced technologies and the implementation of innovations that protect and improve the health of the population. Tackling this challenge requires providing comprehensive support to developers throughout the lifecycle of their innovative products. It is the cooperation of the scientific community, government agencies, and business stakeholders that will smooth the path of medical innovations from the laboratory workbench to the individual patient. In this interview, Tatiana V. Semenova, Deputy Minister of Health of the Russian Federation, describes the most effective support and guidance measures for promising projects in medicine.

INTRODUCTION. The strengthening of the health system and the improvement of access to medicines directly depend on the development and marketing authorisation of national innovative medicinal products. Technology transfer is a key tool in bringing medicinal products to market.
AIM. This study aimed to evaluate the role of the Medical Technology Transfer Centre of the Federal State Budgetary Institution ‘Scientific Centre for Expert Evaluation of Medicinal Products’ of the Ministry of Health of the Russian Federation (FSBI ‘SCEEMP’) in facilitating the implementation of federal projects in medical science and in achieving of pharmaceutical sovereignty.
DISCUSSION. This article is an overview of the main trends in and features of the development of the pharmaceutical industry in the Russian Federation and around the world. The article highlights significant federal decisions made to attain pharmaceutical sovereignty and improve access to medicines for the population. The authors consider documents regulating the work of the Medical Technology Transfer Centre and outline its roles and responsibilities. Additionally, the authors analyse the ability of the Medical Technology Transfer Centre to provide project support throughout the medicinal product development process and to address issues pertaining to efficient pharmaceutical development, patent law, and product commercialisation. The article assesses the role of the Medical Technology Transfer Centre in the development of the pharmaceutical industry in the Russian Federation.
CONCLUSIONS. The Medical Technology Transfer Centre supports projects as part of efforts to implement federal projects in human medical science. The Medical Technology Transfer Centre can be considered a driver of effective innovative development in the national pharmaceutical industry.

INTRODUCTION. The commercial success of innovations in medicine and pharmaceutics depends on the strategic planning of patent protection, with the main elements for consideration being the type of patent protection, the scope of the invention, and the order of obtaining protection documents.
AIM. This study aimed to analyse specific aspects of the strategic planning of patent protection for pharmaceutical inventions.
MATERIALS AND METHODS. The study applied analysis and comparison methods to patent information. Statistical data were prepared using patent databases, including the Rospatent search platform and Questel.
RESULTS. In the Russian Federation, invention patents for methods of treatment and diagnosis are more abundant than pharmaceutical product patents. The most reasonable method for protecting intellectual property rights in the pharmaceutical industry is the stepwise obtaining of different types of protection documents for different objects of patent protection, including inventions, utility models, and industrial designs, as part of one project. The strategy of patent protection for a pharmaceutical invention should consider the type of patent (protection combining the use of patents for inventions, utility models, and industrial designs), the correct determination of the object of patent protection (obtaining a product (substance) patent), and stepwise obtaining and strategic accumulation of different protection documents.
CONCLUSION. The described strategy helps to cover the fullest possible range of rights protection options, prevent patent infringement, and extend the patent protection period for an invention.
INTRODUCTION. The pursuit of technological sovereignty in the current geopolitical situation has led to a growing emphasis on studying relations in the field of government support for healthcare innovation, in particular, the institute of public–private partnership (PPP). In the face of the challenges presented by sanctions, the economic crisis, and a potential deficit in sustainably competitive national innovative medical products, the Russian government is confronted with the necessity of financing the development and production of innovative medical products, including through involving private partners (investors). The use of PPP mechanisms is essential for the achievement of technological sovereignty. In addition to attracting private capital and competences, PPP mechanisms can significantly accelerate the development, realisation, and introduction of innovative medical products, improve the quality of medical care, and increase the production of medicines and medical devices.
AIM. This study aimed to analyse existing PPP mechanisms for the development of the Russian healthcare, science, and technology sectors within the framework of state–business relations and to identify optimal models for the development, realisation, introduction, and production of innovative medical products.
MATERIALS AND METHODS. The study analysed Russian federal legislation using an information-based analytical approach to identify the most promising mechanisms for innovative development in healthcare, with a focus on PPP models.
RESULTS. The article considers frequently encountered forms of possible government involvement in and support of innovative healthcare development with the aim of increasing efficiency and scaling up the realisation and introduction of innovative medical products. Additional aspects covered include the funding of technological sovereignty projects, the expansion of preferential regimes, and the concept of using PPP models. The article presents various PPP realisation mechanisms, including special investment contracts, concession agreements, and PPP agreements, which allow placing infrastructure facilities under the management of private partners. The transferred facilities may include production facilities, research centres, and other facilities necessary for innovative development. The article pays particular attention to the terms and features of concession agreements in the healthcare sector, considers the distribution of duties and responsibilities between the parties involved, and investigates the use of the risk-based approach. Additionally, the article analyses established business practices to identify conditions that are necessary for the parties to a concession agreement.
CONCLUSIONS. The realisation of PPP projects in the innovative development of the healthcare sector requires taking into account and overcoming constraining factors, including technological and administrative barriers that may hinder effective development. The authors believe that the most preferable PPP form is the concession agreement model.
REGULATION OF MEDICINES

INTRODUCTION. The degree of processing (manipulation) of cells included in a cell product and the functions performed after administration (homologous/non-homologous use) determine the classification of the cell product as a transplant or an advanced therapy medicinal product (ATMP) and, hence, the regulatory aspects of the product’s life cycle. Currently, the legislation of the Eurasian Economic Union (EAEU) and the Russian Federation does not sufficiently explain the terms ‘minimal manipulation’ and ‘homologous/non-homologous use’, which may lead to the use of cell products with unproven safety and efficacy in humans.
AIM. This study aimed to compare Russian and international approaches to the interpretation of the terms ‘minimal manipulation’ and ‘homologous/non-homologous use’ for classifying cell products and determining their regulatory pathways, with stromal vascular fraction (SVF) products used as an example.
DISCUSSION. This article reviews and summarises the regulatory approaches of the Russian Federation, the EAEU, the United States (US), and the European Union (EU) that are based on the classification of cell products according to the degree of cell manipulation and the functions performed after administration. The authors have analysed and compared the regulatory acts and approaches of the countries under consideration, with SVF products as a case study. The article highlights general aspects of interpreting the terms ‘minimal manipulation’ and ‘homologous/ non-homologous use’ and demonstrates the difference in regulatory approaches across several countries, which lies in the classification of enzymatic processing and selective collection of cells as substantial or minimal manipulation.
CONCLUSIONS. The mechanism for regulating cell products depends on the degree of cell manipulation (substantial or minimal) and the intended use (homologous or non-homologous). A common principle adopted by regulatory agencies in the US, EU, EAEU, and Russia is to classify manipulation as minimal if the manipulated cells preserve their biological characteristics and physiological function. A defining characteristic of the homologous use of cells or tissues is their administration to perform their inherent functions in the body. In Russia, the regulatory acts for ATMPs and for transplants list the procedures classified as minimal manipulation. According to international standards, preparations based on minimally manipulated SVF cells are classified as ATMPs when used non-homologously. The lack of comprehensive and clear explanations of the terms ‘minimal manipulation’ and ‘homologous/non-homologous use’ in the legislation of the EAEU and the Russian Federation necessitates the development of relevant guidelines providing specific examples.
QUALITY CONTROL
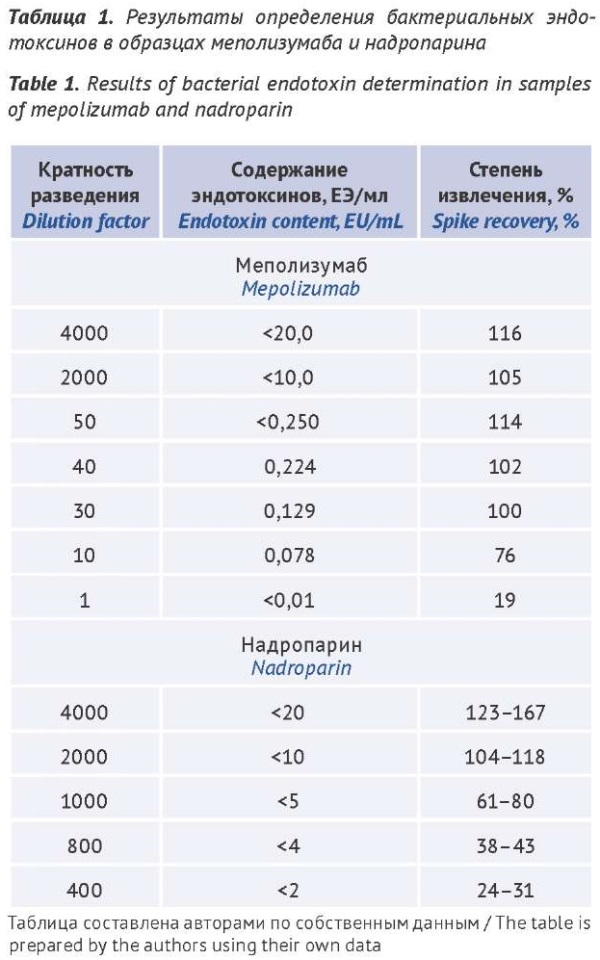
INTRODUCTION. Kinetic bacterial endotoxin (BE) testing methods based on amoebocyte lysate from the haemolymph of the horseshoe crab can quantify BEs in parenteral medicinal products across a broad range of concentrations. To develop a specific analytical procedure that is based on these methods and can be implemented into pharmaceutical quality control and standardisation, it is required to ascertain the conditions for its use, in particular, to determine the lowest product dilution that would neutralise interfering factors.
AIM. This study aimed to develop an analytical procedure for the quantitative determination of BEs by the kinetic chromogenic method.
MATERIALS AND METHODS. The study examined samples of medicinal products containing active substances from different classes, including a monoclonal antibody (mepolizumab) and a direct-acting anticoagulant (nadroparin). The BE limits for mepolizumab and nadroparin were ≤20 EU/mL and ≤95 EU/mL, respectively. The samples were tested at different dilutions, which did not exceed the maximum valid dilution. The measurements were performed using a BioТek EL×800 photocolourimeter and EndoScan-V software.
RESULTS. The results for mepolizumab samples show that the kinetic chromogenic method can quantify BEs in mepolizumab medicinal products in the dilution range of 1:10 to 1:40. The study detected BEs in quantities of 0.0078–0.224 EU/mL within this dilution range. However, at a dilution ratio of 1:50 or higher, the method is suitable only for qualitative BE tests. For nadroparin medicinal products, the quantitative determination of BEs should start at a dilution of 1:1000 (the study demonstrated no influence of interfering factors at this dilution).
CONCLUSIONS. The authors developed analytical procedures for the determination of BEs in mepolizumab and nadroparin medicinal products by the kinetic chromogenic method (method D). When developing analytical procedures for the quantification of BEs in individual medicinal products, the developer should determine the lowest sample dilution that would neutralise interfering factors. Subsequently, the dilution should be specified in the documentation for the analytical procedure. This approach can enhance assay accuracy and save time and resources in routine testing.
INTRODUCTION. Raw materials of biological origin can contaminate medicines with depressor substances, such as histamine, acetylcholine, bradykinin, serotonin, and prostaglandins. In the Russian Federation, tests for the content of these impurities are conducted in anaesthetised healthy cats in accordance with the General Chapter Test for depressor substances of the State Pharmacopoeia of the Russian Federation. The global trend towards ending animal testing and the search for ways to eliminate animal tests for depressor substances motivate researchers to study international approaches to the determination of histamine-like substances.
AIM. This study aimed to analyse Russian and international approaches to the control of biologicals for depressor substances and to consider the applicability of alternative instrument-based physicochemical methods for the determination of histamine-like impurities, with a view to reducing animal testing.
DISCUSSION. The rationale for reducing the use of animals as test systems is set forth in Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes. In 2023, the European Pharmacopoeia Commission initiated the removal of in vivo tests for histamine-like substances from the European Pharmacopoeia. However, China and India use animal testing as the basic method for assessing the safety of medicines. In the Russian Federation, the control of depressor substances is regulated under General Chapter 1.2.4.0008.18 of the State Pharmacopoeia of the Russian Federation. Since the publication of General Chapter 42-0063-07 Test for histamine in edition XII in 2007, the State Pharmacopoeia of the Russian Federation has included a compendial text providing for a reduction in the number of cats used for testing. At present, Russian scientists are searching for targeted physicochemical and immunochemical methods for the determination of depressor substances. It has been demonstrated that a reasonable approach is to assess the need for testing a particular medicinal product in animals on a case-by-case basis. This approach will help minimise the number of in vivo tests and maintain an adequate quality of medicines at the same time.
CONCLUSIONS. Despite the global trend towards ending animal testing, it is still necessary and required to control medicines for the content of depressor substances. Nevertheless, there is no global consensus on the need to use animals for this purpose. The absence of a separate general chapter/monograph, or references to it, will not change the fact that histamine-like substances are controlled worldwide. The decision to exclude in vivo methods from the European Pharmacopoeia necessitates the search for alternative test methods and the implementation of additional manufacturing controls to minimise the contamination risk for finished medicinal products. Studies have shown that high-performance liquid chromatography can determine the content of histamine in a limited amount of a medicinal product.

INTRODUCTION. In 2023, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) published a new Guideline on Analytical Procedure Development (ICH Q14) and a revised version of the Guideline on Validation of Analytical Procedures (ICH Q2(R2)). Consequently, there is a need for a considerable revision of the approach to the development and validation of analytical procedures that is currently used in the Eurasian Economic Union (EAEU). A revision is also needed for the processes for evaluating and introducing variations to the analytical procedures described in medicinal product registration dossiers.
AIM. This review aimed to analyse the significant changes made to international approaches to the development of analytical procedures, as well as to study the advantages and disadvantages of these approaches for pharmaceutical manufacturers and regulatory agencies in the EAEU.
DISCUSSION. This review covers the key provisions and practical aspects of the enhanced approaches to the development of analytical procedures introduced by the ICH Q14 guideline. In particular, the review addresses the concepts of the analytical procedure life cycle (APLC) and the modified analytical quality-by-design (AQbD) approach; the development of the analytical target profile (ATP); analytical quality risk management; planning of the design of experiments (DoE) and the analytical procedure control strategy; and the validation, subsequent verification, transfer, and change management of analytical procedures. Additionally, the review describes the ICH Q2(R2) updates that accompany this new regulatory paradigm.
CONCLUSIONS. The above guidelines fill the existing gap in recommendations for the development of analytical procedures. The use of the APLC and AQbD concepts provides both pharmaceutical companies and regulatory authorities with flexible approaches that are applicable to analytical procedures both during the development phase and once they have been implemented. Effective implementation of these international approaches in the Russian pharmaceutical industry and regulatory system requires a broad discussion between pharmaceutical industry professionals and regulatory agency experts, possibly, as part of a pilot project. After that, there will be a necessity to provide training for specialists involved in the development of analytical procedures and to amend the EAEU Rules for Marketing Authorisation and Expert Assessment of Medicinal Products for Human Use.
PHARMACOPOEIA

INTRODUCTION. Regular reviews of the current portfolio of general chapters and monographs are essential to ensure the well-balanced development of the State Pharmacopoeia of the Russian Federation (Ph. Rus.) in line with the priorities of the national healthcare system.
AIM. This study aimed to identify the most promising areas for further Ph. Rus. development through an analysis of recent legislative changes in Russia and the current compendial portfolio.
DISCUSSION. This article compares Ph. Rus. editions XIV and XV, analyses the key features and structure of edition XV, and reviews amendments to the regulations governing the Ph. Rus. publication. The current compendial portfolio is dominated by monographs on chemical active substances; it also includes a substantial number of monographs on herbal drugs and herbal drug preparations. Noteworthy, edition XV is the first Ph. Rus. edition to include a separate section dedicated to monographs on excipients necessary for the development of pharmacy compounding. Moreover, this article analyses the coverage of active substances that are used for the production of listed vital and essential medicines.
CONCLUSIONS. The authors outlined the priority areas for the development of quality standardisation. The Ph. Rus. should continue developing monographs on active substances that are used for the manufacturing and compounding of listed vital and essential medicines. In addition, the coverage of herbal drugs should be expanded. The Ph. Rus. should develop monographs on excipients that are used for the manufacturing and compounding of medicinal products. Finally, the Ph. Rus. should complete the development of requirements for biological medicinal products and relevant test methods.
HERBAL MEDICINAL PRODUCTS
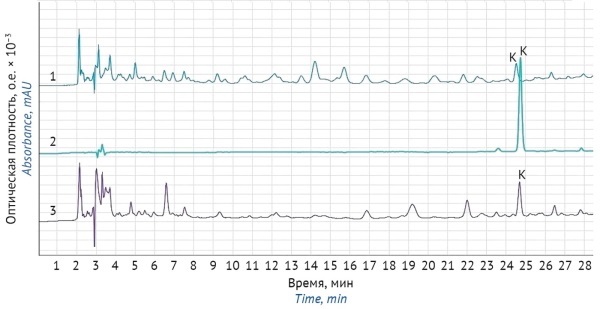
INTRODUCTION. The safe use of lily-of-the-valley medicinal products requires adequately controlling the content of cardiac glycosides. The State Pharmacopoeia of the Russian Federation stipulates that the content of cardiac glycosides in lily-of-the-valley medicinal products should be quantified using bioassays and spectrophotometry. However, the determination of cardiac glycosides needs more accurate and selective physicochemical methods, such as high-performance liquid chromatography (HPLC).
AIM. This study aimed to develop and validate an HPLC analytical procedure for the quantitative determination of cardiac glycosides (convallatoxin) in lily-of-the-valley herbal medicinal products.
MATERIALS AND METHODS. The study examined the Lily-of-the-Valley Tincture, Zelenin Drops, Valocormid, Carniland®, and Lily-of-the-Valley Extract Reference Standard samples. The content of cardiac glycosides was determined by comparison with a reference standard for convallatoxin by HPLC and spectrophotometry. The samples were prepared as outlined in Monograph 3.4.0003.18 Lily-of-the-Valley Herb Tincture of the State Pharmacopoeia of the Russian Federation. Mixed cardiac glycosides were separated on a Luna 5 µm C18(2) column by gradient elution with 0.1% orthophosphoric acid in water and acetonitrile. The analysis was performed using an autoinjector with sample cooling to 5 °C.
RESULTS. The developed analytical procedure met the acceptance criteria for specificity, intermediate precision, linearity (correlation coefficient of 0.99985), and repeatability (relative standard deviation of convallatoxin measurements of 1.61%). The analytical procedure is suitable for the quantitative determination of convallatoxin in lily-of-the-valley herbal medicinal products because it produces reliable and repeatable results.
CONCLUSIONS. The authors developed a highly sensitive and selective HPLC analytical procedure for the quantitative determination of convallatoxin in lily-of-the-valley herbal medicinal products. The content of convallatoxin ranged from 0.012 to 0.018 mg/mL in the liquid active substance Lily-of-the-Valley Tincture, from 0.004 to 0.013 mg/mL in the medicinal product Zelenin Drops, and from 0.005 to 0.007 mg/mL in the medicinal products Carniland® and Valocormid. For the Lily-of-the-Valley Extract Reference Standard, the content of convallatoxin amounted to 0.029 mg/mL.
CLINICAL STUDIES
INTRODUCTION. The examination of protocols for clinical trials of locally applied and locally acting medicinal products highlights challenges that developers face when selecting the design, endpoints, population, comparison groups, and sample size. An analysis of the most common errors in clinical trial protocols will help minimise the number of comments from protocol reviewers and accelerate the process of bringing novel medicinal products to the pharmaceutical market.
AIM. This study aimed to analyse the results of evaluating clinical trials of locally applied and locally acting medicinal products conducted with due consideration of the recent additions to the Rules for Conducting Bioequivalence Studies of Medicinal Products within the Eurasian Economic Union (EAEU); assess the main advantages of the added requirements and the challenges remaining in protocol development; and make recommendations for the most effective application of existing laws and regulations.
DISCUSSION. The Rules for Conducting Bioequivalence Studies of Medicinal Products within the EAEU were supplemented with Appendices 11, 12, and 13 in August 2023. Since then, protocol reviewers have accumulated sufficient experience in the examination of clinical trial protocols for locally applied and locally acting medicinal products. This article presents the most frequent comments made during the examination of clinical trial protocols and provides recommendations for corrective actions. The most challenging aspects of drafting a protocol for a clinical trial of a locally applied and locally acting medicinal product include the selection and justification of primary/secondary endpoints and the calculation and justification of the population size. The difficulty is probably due to the lack of a detailed description of study characteristics in EAEU laws and regulations. Locally applied and locally acting corticosteroids are the least challenging medicinal products in terms of protocol drafting because EAEU legislation and scientific publications provide the most detailed guidance for them.
CONCLUSIONS. The analysis of protocols for clinical trials of locally applied and locally acting medicinal products, relevant EAEU requirements, and applicable international guidelines identified several major challenges, including the selection of primary/secondary endpoints, the characterisation of the trial population, and the justification of the sample size. The recommendations presented in this article will help applicants in planning clinical trials aimed at accelerating the launch of medicinal products into the pharmaceutical market.
INTRODUCTION. Bioanalytical laboratories in the Russian Federation should follow the requirements of both the Good Laboratory Practices (GLP), when performing tests for non-clinical studies, and the Good Clinical Practices (GCP), when analysing samples from clinical studies. The work of such laboratories requires a separate GxP system, the Good Clinical Laboratory Practices (GCLP). However, the GCLP system has not yet been created in the Russian Federation and the Eurasian Economic Union (EAEU).
АIM. This study aimed to compare the current Russian and EAEU principles regulating the work of bioanalytical laboratories with the international GCLP principles and formulate general requirements for national laboratories.
DISCUSSION. The author analysed the current regulation of the work of bioanalytical laboratories, as well as the EAEU regulatory standards for the development and validation of analytical procedures. In addition, the study covered the international GCLP principles that govern the management of human resources, process record keeping, the development of standard operating procedures, the validation of analytical procedures, and the management of biological samples and reference standards in a laboratory. The author considered the key functions of a bioanalytical laboratory and suggested tools to manage them in compliance with the international GCLP principles and the EAEU requirements. It should be noted that scientific publications describe the international practice of applying the GCLP principles fairly well, and the experience of its implementation could be of use to Russian laboratories.
CONCLUSION. A bioanalytical laboratory that implements the GCLP principles will increase its competitiveness in the EAEU market for bioanalytical testing services and mitigate its risks of obtaining invalid data.

ISSN 3034-3453 (Online)





























