


АВТОРИТЕТНОЕ МНЕНИЕ

Конечной целью доклинических исследований является получение данных, которые позволят сделать выводы о возможном воздействии нового лекарственного средства на организм человека и надлежащим образом перейти к клиническим исследованиям. В интервью своими взглядами в отношении проведения доклинических исследований в России поделилась директор АО «НПО «ДОМ ФАРМАЦИИ» доктор медицинских наук Марина Николаевна Макарова.
НОРМАТИВНОЕ РЕГУЛИРОВАНИЕ И РЕКОМЕНДАЦИИ
ВВЕДЕНИЕ. Доклинические исследования (ДКИ) радиофармацевтических лекарственных препаратов (РФЛП) в России и Евразийском экономическом союзе (ЕАЭС) в настоящее время проводятся в соответствии с общими рекомендациями, применимыми для других групп лекарственных препаратов, но не учитывающими особенности РФЛП.
ЦЕЛЬ. Обоснование необходимости разработки методических рекомендаций по проведению ДКИ РФЛП с учетом мировой регуляторной практики.
ОБСУЖДЕНИЕ. Проведен анализ руководств по ДКИ в Российской Федерации и ЕАЭС, зарубежной литературы, а также руководств Управления по контролю за качеством продуктов питания и лекарственных средств (FDA), Европейского агентства по лекарственным средствам (EMA) и Международного агентства по атомной энергии (МАГАТЭ) в части проведения ДКИ РФЛП. Показано, что необходима унификация используемой терминологии. Рассмотрены свойства РФЛП, которые не позволяют при проведении ДКИ использовать рекомендации, применимые для других групп препаратов. Отмечено, что исследования субхронической токсичности терапевтических РФЛП на нерадиоактивном препарате не позволяют оценить вклад излучения радионуклида в биологические эффекты препарата. Предложено проводить изучение системной токсичности готовой лекарственной формы РФЛП по протоколу хронической токсичности с однократным введением.
ВЫВОДЫ. Разработка современных методических руководств по проведению ДКИ РФЛП и их внедрение в практику необходимы для снижения возможных рисков медицинского применения данной группы лекарственных средств. Необходимо учесть различия требований к РФЛП диагностического и терапевтического назначения с учетом правил обеспечения радиационной безопасности. При разработке руководств по ДКИ РФЛП следует ориентироваться на руководства FDA, EMA и МАГАТЭ. Необходимо рассмотреть вопросы о перечне обязательных и об исключении неинформативных исследований из требований к данной группе препаратов.
БЕЗОПАСНОСТЬ ЛЕКАРСТВЕННЫХ СРЕДСТВ
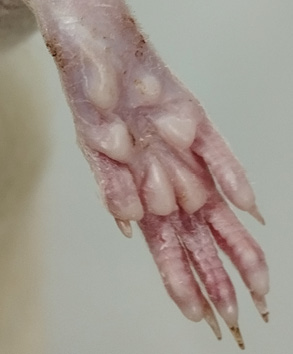
ВВЕДЕНИЕ. В практике доклинических исследований безопасности фармакологически активных веществ стандартные процедуры оценки нейротоксичности в основном нацелены на диагностику расстройств высшей нервной деятельности и поведения. Однако именно структуры периферической нервной системы ввиду большей уязвимости представляют собой доступную мишень, что обусловливает высокую распространенность нейротоксичных побочных эффектов лекарственных средств. Указанные обстоятельства определяют актуальность уточнения методических подходов к оценке токсических поражений периферической нервной системы.
ЦЕЛЬ. Анализ современного методического уровня клинико-функциональных тестов для оценки токсического действия фармакологически активных веществ на структуры периферической нервной системы и формулирование практических рекомендаций по их применению при проведении доклинических исследований на грызунах.
ОБСУЖДЕНИЕ. Оптимальной тест-системой для доклинических исследований фармакологически активных веществ считаются грызуны, однако используя этих животных невозможно воспроизвести весь объем неврологического осмотра, применяемого для выявления клинических эквивалентов нейротоксичности. В работе представлено описание системного подхода к использованию доступных диагностических тестов для повышения транслируемости данных. Дана краткая характеристика неврологического дефицита, вызванного побочными действиями лекарственных средств у людей, и описаны основные токсиндромы, которые также могут быть выявлены у животных. На основании обзора литературы и собственного опыта в соответствующих разделах представлены практические рекомендации по выполнению основных тестов, позволяющих исследовать силу и тонус мышц, состояние физиологических рефлексов, координацию движений и разные виды чувствительности у грызунов. Приведены краткие сведения о возможностях электрофизиологической диагностики искомых поражений. В качестве минимального перечня методик первичного скрининга нейротоксических побочных эффектов рекомендованы следующие тесты: оценка позы животного в покое и при ходьбе «Сужающаяся дорожка», «Подтягивание на горизонтальной перекладине», шкала отведения пальцев, «Тест отдергивания хвоста», рефлекс Прейера.
ВЫВОДЫ. Анализ результатов комплексной оценки неврологического дефицита в экспериментах на грызунах рекомендовано проводить клинически релевантным способом, то есть с позиций топической диагностики и общности звеньев патологического процесса. Целесообразно выполнять верификацию патологического процесса на уровне периферической нервной системы при помощи комплекса электрофизиологических методик.
ВВЕДЕНИЕ. Оценка функции мочевыделительной системы, и в большей степени почек, является важной задачей доклинических исследований. В настоящее время не существует общепризнанного и детального подхода к определению лекарственно-индуцированной нефротоксичности in vivo и четких критериев ее оценки.
ЦЕЛЬ. Систематизация инструментальных и лабораторных методов оценки функций мочевыделительной системы лабораторных животных и выявление основных принципов изучения нефротоксического действия лекарственных средств.
ОБСУЖДЕНИЕ. Проанализированы преимущества и ограничения методов исследования нефротоксичности лекарственных средств. Рассмотрены особенности их применения у мелких и крупных лабораторных животных. Целесообразно начинать оценку влияния веществ на мочевыделительную систему с малоинвазивных методов. Один из таких методов — анализ мочи, важными аспектами которого являются техника отбора и объем биоматериала, а также временной интервал между забором мочи и проведением теста. Наиболее доступным инструментальным методом в рамках доклинических исследований является ультразвуковое исследование, которое позволяет оценить положение органов, их размеры, структуру и эхогенность, обнаружить аномалии и изменения в режиме реального времени. Для каждого лабораторного вида предпочтительны свои настройки метода. К последующим этапам относятся макроскопическое изучение органов, измерение их массы и микроскопический анализ тканей. Визуально необходимо оценивать размеры, цвет и консистенцию мочеточников, мочевого пузыря и почек. Нефротоксичность может быть обнаружена в виде усиленного апоптоза, вакуолизации цитоплазмы эпителия почечных канальцев, дегенерации или дистрофии эпителия, отека, диапедезных кровоизлияний, острого канальцевого и папиллярного некроза, некроза капсулы Боумена — Шумлянского, возникновения слепков и кристаллов в просвете канальцев, развития гломерулопатий с соответствующими изменениями, а также воспалительных и сосудистых реакций.
ВЫВОДЫ. В результате анализа и систематизации инструментальных и лабораторных методов оценки функционального состояния мочевыделительной системы в доклинических исследованиях были обозначены основные принципы структурированного и всестороннего изучения потенциальной нефротоксичности новых лекарственных средств. Оценку нефротоксичности целесообразно начинать с простых и малоинвазивных лабораторных и инструментальных методов, к которым относится общий анализ мочи и микроскопия мочевого осадка, что позволяет определить наличие нарушения функции органа, еще не имеющего сопутствующего анатомического поражения. При более глубоком анализе следует использовать методы гистологического и иммуногистохимического исследований тканей органов мочевыделительной системы животных.
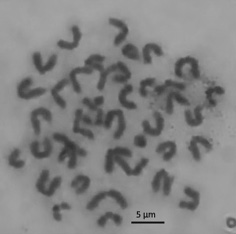
ВВЕДЕНИЕ. Для определения мутагенности лекарственных средств используется метафазный анализ, точность теста напрямую зависит от качества приготовленных цитогенетических препаратов. Возможная артефактная потеря хромосом, приводящая к ошибкам при трактовке результатов, длительность и трудоемкость пробоподготовки обусловливают необходимость совершенствования существующих методик подготовки цитогенетических препаратов.
ЦЕЛЬ. Оптимизация методики приготовления цитогенетических препаратов клеток костного мозга млекопитающих in vivo для метафазного анализа.
МАТЕРИАЛЫ И МЕТОДЫ. Исследования выполнены на беспородных мышах-самцах массой 18–20 г. Пробоподготовку цитогенетических препаратов костного мозга выполняли по оригинальным и адаптированным методикам «Руководства по проведению доклинических исследований лекарственных средств» под ред. Миронова А.Н. (2012 г.) и ГОСТ 34659-2020 «Методы испытания по воздействию химической продукции на организм человека. Оценка хромосомных аберраций в клетках костного мозга млекопитающих». Для приготовления препаратов использовали раствор Хенкса, растворы цитрата натрия и хлорида калия, уксусную кислоту, метиловый спирт, среду 199, буферный раствор HEPES и краситель Гимзы.
РЕЗУЛЬТАТЫ. Проведен сравнительный экспериментальный анализ характеристик цитогенетических препаратов для проведения метафазного анализа клеток костного мозга млекопитающих in vivo, подготовленных по различным методикам. Выявлено, что препараты, полученные по методике «Руководства по проведению доклинических исследований лекарственных средств», характеризовались максимальной долей доступных для анализа пластинок по сравнению с препаратами, полученными другими способами пробоподготовки. Оптимизированы условия пробоподготовки, в том числе состав гипотонического раствора (0,56% раствора KCl), время гипотонической обработки (20 мин) и фиксации (12 ч), состав культуральной среды (среда 199 жидкая с солями Хенкса с глутамином), введена стадия префиксации хромосом 6%-ным раствором уксусной кислоты, что в целом способствовало увеличению доли доступных для анализа метафазных пластинок путем снижения разброса и слипания хромосом.
ВЫВОДЫ. Предложена методика приготовления цитогенетических препаратов, позволяющая увеличить долю доступных метафазных пластинок для анализа до 56%, долю хромосом с полным набором (n=40) на 12% и сократить время приготовления образцов на 2–10 ч по сравнению с использовавшимися ранее методиками.
БИОАНАЛИТИЧЕСКИЕ МЕТОДЫ
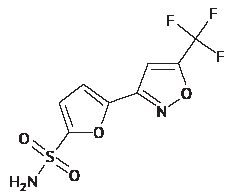
ВВЕДЕНИЕ. Для изучения системной экспозиции селективного ингибитора карбоангидразы II типа производного изоксазола 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамид (TFISA) необходима оценка его фармакокинетических параметров в цельной крови, поскольку это соединение способно накапливаться в эритроцитах. Ранее биоаналитические методики для решения данной задачи разработаны не были.
ЦЕЛЬ. Разработка биоаналитической методики определения 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида и его метаболитов N-гидрокси-5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида и N-ацетил-5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида в крови лабораторных животных и сравнение фармакокинетики глазной суспензии TFISA после однократной инстилляции и внутрибрюшинного введения крысам.
МАТЕРИАЛЫ И МЕТОДЫ. Количественное определение проводили методом высокоэффективной хроматографии с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС) с использованием образцов крови крыс и кроликов. Хроматографическое разделение осуществляли с помощью колонки Zorbax Eclipse Plus C18 (150×3,0 мм, 3,5 мкм) c применением 0,1% водного раствора муравьиной кислоты и метанола для градиентного элюирования. Масс-спектрометрическое детектирование выполняли в режиме мониторинга множественных реакций. Изучение фармакокинетики проводили на двух группах крыс линии Wistar по 6 особей (по 3 самца и 3 самки). Первой группе животных осуществляли инстилляцию 1% глазной суспензии TFISA в каждый глаз из расчета 3,7 мг/кг. Животным второй группы вводили этот же препарат внутрибрюшинно в той же дозе. Образцы крови отбирали до введения препарата, а также спустя определенные временные интервалы после введения.
РЕЗУЛЬТАТЫ. Разработана ВЭЖХ-МС/МС-методика количественного определения TFISA и его метаболитов в крови лабораторных животных (кроликов и крыс). ВЭЖХ-МС/МС-методика полностью валидирована в соответствии с нормативными актами Евразийского экономического союза и требованиями руководства ICH M10. Аналитический диапазон определения TFISA в крови составил 20–20000, N-гидроксипроизводного — 2–2000, N-ацетилпроизводного — 0,1–100,0 нг/мл. Максимальная концентрация TFISA в крови после инстилляции в глаз достигала 8173±1491, N-гидроксипроизводного — 695±271, N-ацетилпроизводного — 6,33±1,51 нг/мл. Период полувыведения TFISA при данном пути введения составил 58±10, N-гидроксипроизводного — 70±24, N-ацетилпроизводного — 14±3 ч. Биодоступность действующего вещества составила 90,18%. ВЫВОДЫ. Разработанная биоаналитическая методика определения 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида (TFISA) и его метаболитов в крови лабораторных животных была успешно использована для анализа образцов цельной крови крыс. В ходе исследования фармакокинетики глазной суспензии данного соединения выявлен длительный период полувыведения действующего вещества и его метаболитов, а также высокая относительная биодоступность.
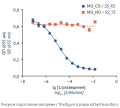
ВВЕДЕНИЕ. Для биоаналитических методик характерны бо́льшая вариабельность и меньшая устойчивость по сравнению с физико-химическими методиками в силу лабильности характеристик живых тест-систем. Поскольку в нормативных документах не определен единый подход к формированию перечня критериев пригодности системы и допустимых границ для полученных результатов, то их устанавливают на основании валидационных испытаний.
ЦЕЛЬ. Получить экспериментальное подтверждение соответствия методики оценки биологической активности лекарственного средства на основе разрабатываемого биоаналога тоцилизумаба (GNR-087) валидационным требованиям и определить количественные границы критериев пригодности аналитической системы и приемлемости результатов анализа.
МАТЕРИАЛЫ И МЕТОДЫ. Биологическую активность биоаналога тоцилизумаба оценивали по ингибированию интерлейкин-6-зависимой секреции эмбриональной щелочной фосфатазы (SEAP), продуцируемой клетками репортерной линии HEK-Blue™ IL-6. Статистическую обработку полученных результатов проводили с использованием программного обеспечения Prism 6.0.
РЕЗУЛЬТАТЫ. Специфичность методики подтверждена дозозависимым ингибированием интерлейкин-6-зависимой секреции эмбриональной щелочной фосфатазы клетками при действии тоцилизумаба. Методика линейна, коэффициент детерминации линейной функции R2≥0,99. Прецизионность методики удовлетворительная, сходимость — от 2 до 9%, внутрилабораторная прецизионность — 9 и 14%. Коэффициенты выявления активности (Rc) модельных образцов с симулированной активностью от 60 до 140%, в том числе и «слепых проб», находились в диапазоне 80–120%. Теоретические значения относительной специфической активности (RP) находились внутри доверительных интервалов средних найденных значений RP, что подтвердило правильность методики. Подтверждена устойчивость методики к контролируемым изменениям: использованию репортерных клеток разных пассажей (коэффициент вариации (CVRP) составил 10%), замене лотов ИЛ-6 (CVRP=1%) и реагента для детекции SEAP (CVRP=3%), при этом значение Rc находилось в диапазоне 80–120% от номинального значения RP.
ВЫВОДЫ. Методика оценки биологической активности биоаналога тоцилизумаба соответствует валидационным характеристикам, таким как правильность, линейность, прецизионность, специфичность и устойчивость к контролируемым изменениям. Установлены границы критериев пригодности системы и рассчитаны критерии приемлемости результатов биологического анализа. Разработанная методика может служить как для рутинного контроля биологической активности, так и для использования в исследованиях при доказательстве биоподобия разрабатываемых препаратов оригинальному (референтному) препарату, содержащему тоцилизумаб.

ВВЕДЕНИЕ. Тенденция к сокращению использования лабораторных животных при оценке качества лекарственных препаратов, а также появление новых технологий получения гонадотропных препаратов обусловливают необходимость разработки новых методик определения биологической активности природных и рекомбинантных препаратов передней доли гипофиза.
ЦЕЛЬ. Выбор оптимальных условий определения биологической активности лютеинизирующего гормона в мочевых и генно-инженерных препаратах в испытаниях на крысах беспородных и линии Sprague Dawley.
МАТЕРИАЛЫ И МЕТОДЫ. Определение величины биологической активности мочевых и рекомбинантных препаратов лютеинизирующего гормона (ЛГ) проводили трехдозовым рандомизированным методом in vivo по схеме Steelman and Pohley путем сравнения биологической активности испытуемых и стандартных (СО) образцов. Для анализа были использованы результаты, полученные в течение двух лет. В качестве СО использовали стандарт ВОЗ, содержащий 183 МЕ фолликулостимулирующего гормона и 177 МЕ ЛГ/амп. Испытание проводили на неполовозрелых крысах-самцах беспородных и линии Sprague Dawley. В зависимости от статуса крыс выбирали условия проведения испытания.
РЕЗУЛЬТАТЫ. Проведен сравнительный анализ результатов определения активности ЛГ in vivo на крысах-самцах по интегральному показателю s/b, позволяющему оценить одновременно разброс ответов и дозозависимость. Было показано, что использование самцов крыс линии Sprague Dawley (s/b=0,82±0,44) для определения биологической активности ЛГ возможно наравне с беспородными животными (s/b=0,68±0,58). Значения показателей s/b близки, несмотря на то что количество животных линии Sprague Dawley в испытаниях было в два раза меньше.
ВЫВОДЫ. Показана возможность определения биологической активности ЛГ в препаратах менотропинов не только на беспородных, но и на линейных крысах-самцах. Проведение испытания на крысах линии Sprague Dawley позволяет подтвердить достоверность результатов, используя меньшее количество животных, что, в свою очередь, является не только гуманным, но и более выгодным экономически.
КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
ВВЕДЕНИЕ. Корректное планирование клинического исследования (КИ) является гарантией получения валидных результатов оценки эффективности и безопасности медицинского применения лекарственных средств. В настоящее время отсутствуют четкие критерии выбора базовых элементов, лежащих в основе разработки клинического дизайна, и прежде всего исследовательских гипотез, способов определения ожидаемой величины терапевтического эффекта, уровня статистической значимости и мощности исследования, статистических моделей расчета размера выборки субъектов.
ЦЕЛЬ. Систематизация и гармонизация технических требований к планированию дизайна клинического исследования в части определения размера выборки.
ОБСУЖДЕНИЕ. В работе представлены основные требования и методологические подходы к разработке дизайнов медицинских исследований, направленных на оценку эффективности и подтверждение безопасности лекарственных средств. Приведены базовые принципы расчета необходимого размера выборки для обеспечения необходимой мощности планируемого КИ, а также математические модели, описывающие нулевые и альтернативные гипотезы, используемые при разработке основных статистических дизайнов исследования эффективности и безопасности лекарственных препаратов. Показано, что общим требованием к качеству выборки субъектов исследования является обеспечение ее репрезентативности, то есть соответствие целевой популяции КИ. Выбор математической (вероятностной) модели, на основе которой формулируются исследовательские гипотезы и производится расчет выборки целевой популяции, базируется прежде всего на базовой информации о терапевтическом воздействии и специфических особенностях популяции, полученной из систематических обзоров результатов ранее проведенных исследований, а также в соответствии с классификацией исследуемого препарата. Для расчета размера выборки должны быть определены и обоснованы на этапе разработки дизайна и статистической модели КИ критерии в соответствии с общими требованиями к репрезентативности. Использование программных приложений для расчета мощности и требуемого размера выборки упрощает выполнение рутинных процедур планирования клинических исследований.
ВЫВОДЫ. Основных и базовых статистических моделей определения размера выборки недостаточно для проведения качественного исследования. Большое разнообразие дизайнов КИ, методологических подходов к планированию, реализации схем лечения, сбора и анализа данных КИ требует разработки статистических планов каждого конкретного КИ, включая оценку отдельных случаев, метода анализа выживания, относительного риска, диагностические тесты, адаптивные и другие нечасто используемые планы исследования. Следствием этого является востребованность в разработке дополнительных руководств и других информационных ресурсов, содержащих комментарии и примеры применения вероятностной статистики, и последующей гармонизации созданных национальных стандартов с международными.
ВВЕДЕНИЕ. Генерализованное тревожное расстройство (ГТР) является наименее изученным из ряда тревожных расстройств ввиду наличия у пациентов сопутствующей патологии, связанной с расстройствами настроения. Поиск эффективных методов лечения ГТР является крайне важной задачей, что и обусловливает актуальность разработки новых лекарственных препаратов для лечения ГТР. Надлежащее планирование программы проведения клинических исследований является залогом получения достоверных данных об эффективности и безопасности лекарственных препаратов. В настоящий момент методическое руководство по проведению клинических исследований ГТР в Российской Федерации отсутствует и поставлена задача его разработки.
ЦЕЛЬ. Оценка возможности использования методологических подходов зарубежных руководящих документов к проведению клинических исследований лекарственных средств в российской клинической практике при разработке лекарственных средств для терапии генерализованного тревожного расстройства.
ОБСУЖДЕНИЕ. Выполнен анализ основных положений Руководства по клиническим исследованиям препаратов для терапии ГТР Европейского агентства по лекарственным средствам (European Medicines Agency, EMA). Описаны основные этапы клинических исследований лекарственных препаратов и методология использования результатов этих исследований для оценки эффективности и безопасности данной группы препаратов. Показано, что при разработке программы клинических исследований ГТР необходимо учитывать этапность проведения исследований, обязательной является долгосрочная оценка безопасности и аддитивного эффекта. Выделены аспекты выбора дизайна клинических исследований, формирования групп исследований, а также определения первичных и вторичных конечных точек исследований. Особый акцент сделан на учете коморбидности пациентов.
ВЫВОДЫ. Положения руководства ЕМА могут послужить основой отечественного руководства по изучению лекарственных препаратов для лечения генерализованного тревожного расстройства.
МИКРОБИОЛОГИЧЕСКИЙ АНАЛИЗ
ВВЕДЕНИЕ. Первым этапом микробиологического анализа лекарственного средства (ЛС) является оценка его антимикробного действия, то есть влияния ЛС на рост аэробных бактерий, дрожжевых и плесневых грибов. Совершенствование методических инструментов для проведения микробиологического анализа с целью повышения качества получаемых данных остается актуальной задачей.
ЦЕЛЬ. Разработка методического подхода к определению микробиологической чистоты нестерильных лекарственных средств, обладающих антимикробным действием, повышение точности и надежности существующих методик микробиологического анализа.
МАТЕРИАЛЫ И МЕТОДЫ. Для исследования использовали нестерильные ЛС 12 наименований, 5 разбавителей (фосфатный буферный раствор, фосфатный буферный раствор с добавкой полисорбата-80 (1 или 5%), нейтрализующая жидкость, триптиказо-соевый бульон), питательные среды и тест-микроорганизмы, регламентированные Государственной фармакопеей Российской Федерации и Фармакопеей Евразийского экономического союза для проведения микробиологического анализа качества.
РЕЗУЛЬТАТЫ. Определены условия проведения микробиологического исследования нестерильных ЛС с выявленным антимикробным действием, разработанные методики апробированы на ЛС 10 наименований. Доказана применимость методик количественного определения микроскопических грибов в образцах с фунгистатическим действием. Коэффициенты прорастания Candida albicans и Aspergillus brasiliensis находились в допустимом диапазоне 76–128%. Предложено внесение изменений в методику качественного определения Escherichia coli: разведение образца 1:50 в количестве 50 мл (соответствующее 1 г ЛС), которое вносили в 450 мл соответствующей питательной среды. Сравнение разбавителей, содержащих в составе неспецифические инактиваторы антимикробного действия, показало, что предпочтительным является использование нейтрализующей жидкости.
ВЫВОДЫ. Предложен методический подход к определению микробиологической чистоты ЛС, основанный на том, что результаты анализа следует считать достоверными только с учетом антимикробного действия испытуемого образца. Доказана целесообразность увеличения количества образца до 10 мл из разведения, в котором антимикробное действие отсутствует (например, 1:100). Для посева рекомендовано использовать чашки Петри диаметром 150 мм, 50 мл агаризованной питательной среды, а также разбавители, содержащие в составе до 5% полисорбата-80. Показана применимость разведения, при котором антимикробное действие отсутствует; необходимость увеличения количества образца пропорционально вносимому в питательную среду до регламентированного фармакопеями количества — не меньше 1 г (мл), а также рекомендовано использовать разбавители, содержащие в составе до 5% полисорбата-80, для инактивации бактериостатического и фунгистатического действия.
ЭКСПЕРТНОЕ МНЕНИЕ
Предложены рекомендации по изложению в нормативном документе (нормативной документации) методики оценки качества лекарственных средств по показателю «Определение воды» при использовании метода К. Фишера. Рассмотрены особенности проведения анализа и учета результатов, предложена типовая схема редакционного оформления раздела. Унификация изложения и оформления текста нормативной документации позволит безошибочно выполнять испытание, получать достоверный результат и упростит экспертизу лекарственных средств.

ISSN 3034-3453 (Online)





























