


MAIN TOPIC: INNOVATION IN PHARMACEUTICS: FROM SCIENTIFIC IDEA TO TECHNOLOGICAL LEADERSHIP

In this interview, D.I. Fedorova, First Deputy Director of the Medical Technology Transfer Centre at the Scientific Centre for Expert Evaluation of Medicinal Products, describes practical, data-driven tools that support the management of scientific and technological development in the pharmaceutical sector. The central topic is technology mapping, an approach that enables a systems-level analysis of research directions, technology maturity levels, and implementation potential. The interview outlines how this tool can be used by government authorities and other decision-makers to adjust research priorities, allocate resources, and mitigate risks, and what value it provides to pharmaceutical and biotechnology companies when building development portfolios.

INTRODUCTION. Advances in gene therapy technologies and their expanding clinical use are progressing faster than the evolution of regulatory frameworks. In the Russian Federation, oversight of cell- and gene-based therapies remains complex and fragmented due to the interplay between national legislation and supranational regulations within the Eurasian Economic Union.
AIM. This study aims to examine the current regulatory framework governing the lifecycle of gene therapy products in the Russian Federation, including their authorization, manufacturing, and funding; to identify regulatory gaps and inconsistencies; and to propose directions for policy improvement.
MATERIALS AND METHODS. A combined systems-based and problem-oriented approach was applied to the analysis of regulatory legal acts. The study was conducted in four stages: first, a content analysis of the regulatory framework was performed to identify key concepts; second, requirements across different legal acts were compared to detect areas of ambiguity and regulatory inconsistency; third, the identified issues were systematized using structural-functional and logical analysis; and finally, recommendations were formulated based on the integrated findings.
RESULTS. The analysis covered Federal Laws No. 61 and No. 180, decisions of the Eurasian Economic Commission Council, and subordinate regulations governing Good Manufacturing Practice (GMP) and the State Guarantees Program (SGP). Depending on their characteristics, gene therapy products may fall under several regulatory categories, including advanced therapy medicinal products (ATMPs), biotechnological medicinal products, or biomedical cell products, each associated with distinct requirements for market authorization, manufacturing, and patient access. The study highlights fundamental differences in the regulatory treatment of personalized therapies compared with standardized products. Notably, there is currently no established legal framework permitting centralized, industrial-scale manufacturing of personalized therapies with subsequent transport of biological materials, which poses a significant barrier to scalability. In addition, further development of reimbursement mechanisms is needed for gene therapy interventions that do not require market authorization, in order to broaden patient access beyond those treatments already covered by the SGP, specifically within List 4 of High‑Tech Medical Care.
CONCLUSIONS. Improving access to innovative therapies will require alignment of Federal Laws No. 61 and No. 180 with Decision No. 78 of the Eurasian Economic Commission Council, harmonization of classification criteria for gene therapy products, establishment of a regulatory pathway for centralized manufacturing of personalized therapies, and the introduction of more flexible reimbursement models to support patient access.
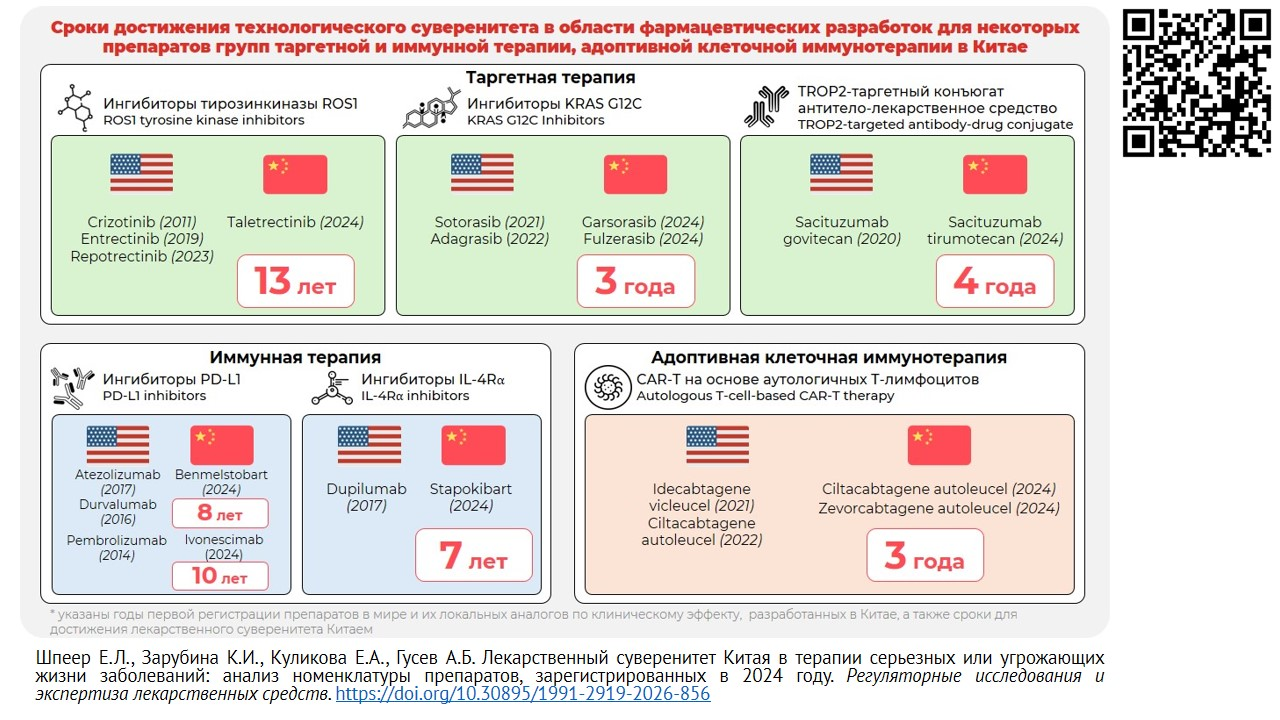
INTRODUCTION. The policy of friendly states aimed at achieving and maintaining the state sovereignty regarding drug safety is essential when forming Russian strategy of pharmaceutical development. Considering rapid advancement of biomedical technologies and the growing role of national regulatory frameworks, China’s experience in developing and launching innovative medicinal products to the local market is of interest.
AIM. This study aimed to analyze the list of Class 1 medicinal products classified by National Medical Products Administration of China (NMPA) and developed by Chinese companies to treat serious and life-threatening conditions and registered in China in 2024, focusing on their potential first-in-class status, degree of innovation, key technological solutions, and potential clinical significance.
MATERIALS AND METHODS. Medicinal products developed in China within the breakthrough therapy program and registered in 2024 according to NMPA data were assessed. The analysis included the 2024 NMPA report.
RESULTS. Nine medicinal products first approved in China and developed by Chinese companies were identified that were registered as Class 1 medicinal products aimed at treating serious or life-threatening conditions. These included targeted therapies, immunotherapies, antibody–drug conjugates, and CAR-T cell therapies developed by Chinese manufacturers. For each product, the mechanism of action, process cha racteristics, availability of similar foreign preparations, and clinical evidence from approval trials were examined. One medicinal product (ivonescimab) may be classified as first-in-class, whereas the remaining products were categorized as next-in-class. For several products (ivonescimab, taletrectinib, and ciltacabtagene autoleucel), post-registration data indicated a potential best-in-class profile. The analysis highlights that the key process strategies were primarily aimed at enhancing target selectivity, overcoming resistance mechanisms, optimizing pharmacokinetic properties, and improving safety profiles.
CONCLUSIONS. The findings demonstrate the emerging mature and structured model for the accelerated development of innovative medicinal products in China, characterized by a high potential for clinical impact. The obtained results may be of interest to Russian developers considering the current national programs aimed at achieving technological sovereignty in pharmaceutical development.

INTRODUCTION. Despite the widespread use of solid dosage forms (SDFs) with modified release (MR), there is a paucity in the literature of comprehensive reviews covering the full development cycle — from selection of a technological platform to the application of digital optimization tools. Existing publications typically focus on individual technological platforms without addressing digital optimization.
AIM. To systematically review and critically evaluate modern approaches to optimi zing the manufacturing technology for obtaining of modified-release solid dosage forms, and to determine the role of digital tools in enhancing the efficiency of pharmaceutical development.
DISCUSSION. The analysis of scientific publications showed that hydrophilic matrices based on hydroxypropyl methylcellulose (HPMC) remain the leading platform for the development of prolonged-release formulations. However, controlling the release of highly soluble drugs requires combining hydrophilic and hydrophobic polymers. Osmotic systems provide a pH-independent release profile, but their use is associated with the risk of gastrointestinal irritation. Multiparticulate forms reduce the likelihood of unintentional release of dose dumping; in such systems, the quality of the polymer film coating governs the reproducibility of release kinetics. Hot-melt extrusion combined with 3D printing using fused deposition modeling (FDM) enables the creation of a desired release profile by varying the geometry of the dosage form. It has been established that the Quality by Design concept and machine lear ning methods substantially reduce the experimental workload, although challenges related to the interpretability of deep neural networks and the lack of external validation of predictive models remain unresolved.
CONCLUSIONS. Matrix systems based on HPMC hold a leading position among prolonged-release technologies owing to their scalability and predictable kinetics; however, the optimal choice of technology for solid dosage forms with modified release is determined by the physicochemical properties of the substance and the pharmacokinetic profile. The application of design of experiments methods and machine learning in the development of MR SDFs can substantially reduce the number of laboratory experiments. A promising direction for future research lies in the development of interpretable predictive models and the adaptation of the regulatory framework to dosage forms produced by additive manufacturing (three-dimensional printing).
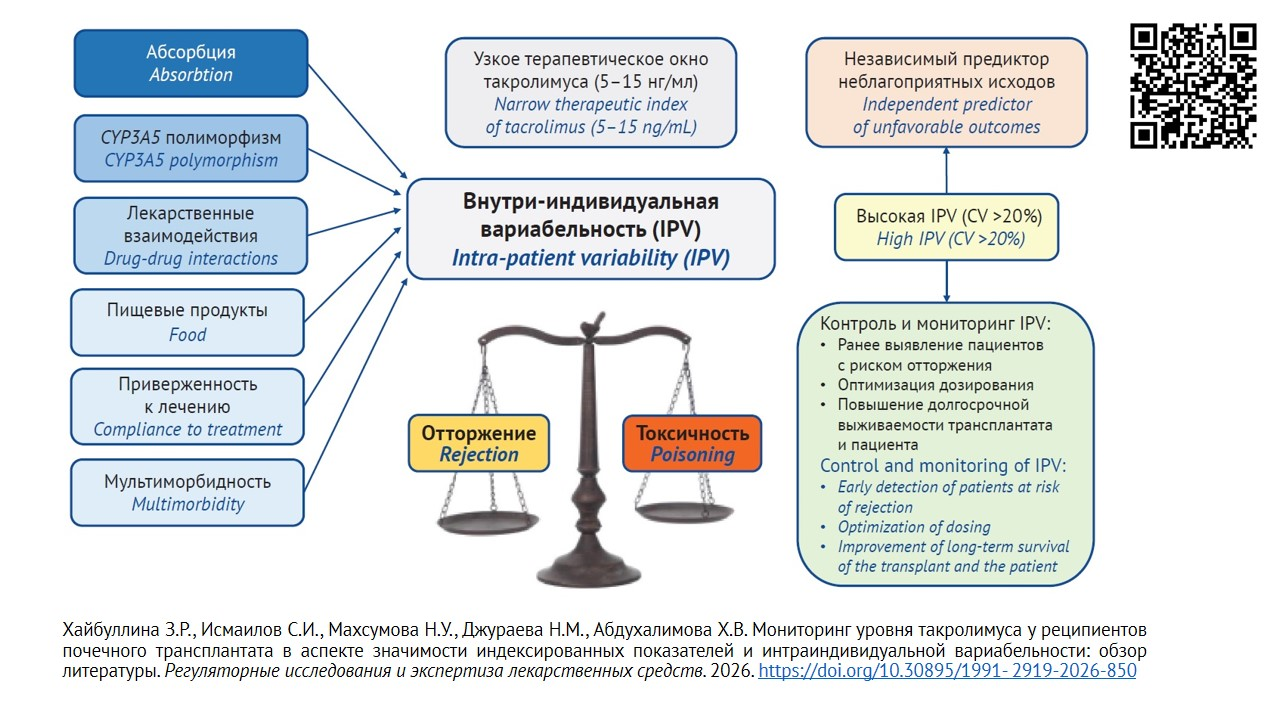
INTRODUCTION. Tacrolimus (TAC) in combination with mycophenolate mofetil and glucocorticoids is a cornerstone of immunosuppressive therapy after kidney transplantation. The analytical sensitivity of methods used to determine TAC concentrations in blood samples varies widely and associated with the risk of transplant rejection that requires regular monitoring.
AIM. To evaluate methods for measuring tacrolimus blood concentrations in kidney transplant recipients and compare different approaches to monitoring tacrolimus exposure.
DISCUSSION. Liquid chromatography with mass spectrometry (LC/MS/MS) is the standard for quantitatively assessing TAC in various biomaterials. Immunochemical methods for TAC determination in whole blood have been introduced into clinical practice; however, due to cross-reactivity with inactive TAC metabolites, the results may be overestimating. Immunochemiluminescence method, using magnetic particles, ensures reliable results and allows to determine TAC concentrations down to 0.5 ng/mL, while the coefficient of variation values does not exceed 15% compared to the reference LC/MS/MS method. TAC has a narrow therapeutic window, and its levels in kidney transplant recipients, both in whole blood / plasma and in mononuclear cells, are characterized by high intra-patient variability (IPV). It necessitates the development of new approaches to assessing target TAC levels, including the choice of biomaterial and the method for TAC determination in patients with risk of kidney transplant dysfunction. The most clinically significant marker of immunosuppressive efficacy is the steady-state concentration of TAC in the blood. The C/D ratio has been proposed for toxicity prediction: a decrease in C/D ratio may be a predictor of an unfavorable prognosis due to TAC toxicity in kidney transplant recipients. The IPV directly reflects the stability of drug exposure in a given patient and allows for assessing the risk of transplant rejection and toxicity. A high IPV is an independent predictor of adverse outcomes in kidney transplant recipients. The IPV of TAC concentration depends on endogenous and exogenous factors, such as CYP3A5 polymorphism, dietary factors, drug-drug interactions, and clinical situations. Regular monitoring of IPV and elimination of influencing factors help ensure both immediate and long-term good survival of transplants.
CONCLUSIONS. Tacrolimus has a narrow therapeutic window, and its blood levels in kidney transplant recipients exhibit high IPV. Inter-laboratory comparison and the development of normalized values will minimize variability in TAC concentration assessments, while accounting for IPV will reduce the risk of adverse events during short- and long-term follow-up of kidney transplant recipients. This justifies a revision of approaches to monitoring TAC concentrations to improve treatment outcomes and increase kidney transplant and recipient survival.
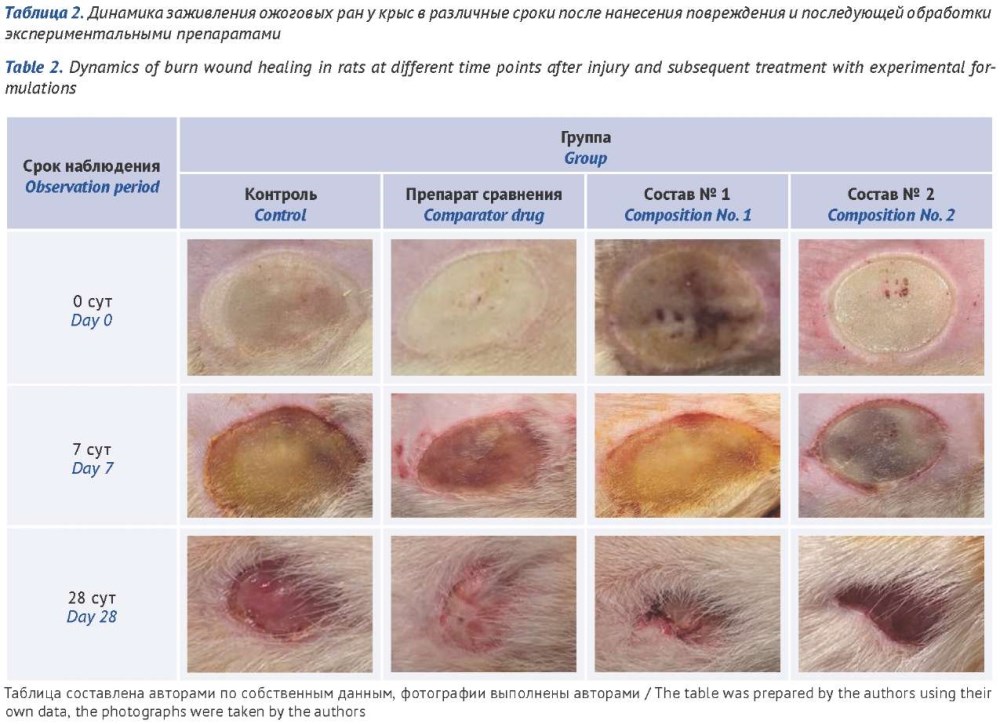
INTRODUCTION. Implementation of the state strategy “Pharma 2030” requires the development of high-tech domestic drugs, including acellular tissue-engineered composites characterized by high efficacy and low immunogenicity. As part of this effort, a regenerative spray for the treatment of grade IIb–IIIa burn wounds is being developed and its pharmacological properties are being studied. This spray is a tissue-engineered composite of acellular human umbilical cord biomaterial and an antimicrobial component.
AIM. To evaluate the regenerative activity of experimental spray formulations derived from human umbilical cord Wharton’s jelly in a rat burn model.
MATERIALS AND METHODS. The developed spray formulations contained a biodegradable lyophilized hydrolysate of the acellular matrix of human umbilical cord Wharton’s jelly and antibacterial agents: formulation No. 1 included gentamicin sulfate, formulation No. 2 included neomycin sulfate. Olazol aerosol (Altayvitamins, Russia) was used as the reference drug. The study was performed on a deep burn model in adult rats. The animals were divided into groups that received treatment with the experimental sprays, the reference drug, a control group (no treatment), and a group of intact animals. Regenerative activity was assessed by measuring the wound defect area and by determining blood leukocyte count, C-reactive protein (CRP) concentration, and epidermal growth factor (EGF) level.
RESULTS. By day 28 of observation, both spray formulations showed a statistically significant advantage over the reference drug in reducing the burn wound area: formulation No. 1 by 66%, formulation No. 2 by 20%. Treatment with the experimental formulations and the reference drug resulted in a decrease in leukocyte count and CRP level to values comparable with those of intact animals. The blood EGF concentration increased significantly in the experimental groups: with formulation No. 1 by 104.0% and with formulation No. 2 by 76.5% compared with the control group, and by 106.5% and 78.8%, respectively, compared with the intact animal group.
CONCLUSIONS. The experimental spray formulations derived from human umbilical cord Wharton’s jelly demonstrate high regenerative activity, significantly accelerating the healing of deep burn wounds and reducing the severity of the inflammatory response in rats. The results support the feasibility of further preclinical and clinical investigation of these compositions, with a view to their introduction into clinical combustiology.
QUALITY CONTROL OF MEDICINES
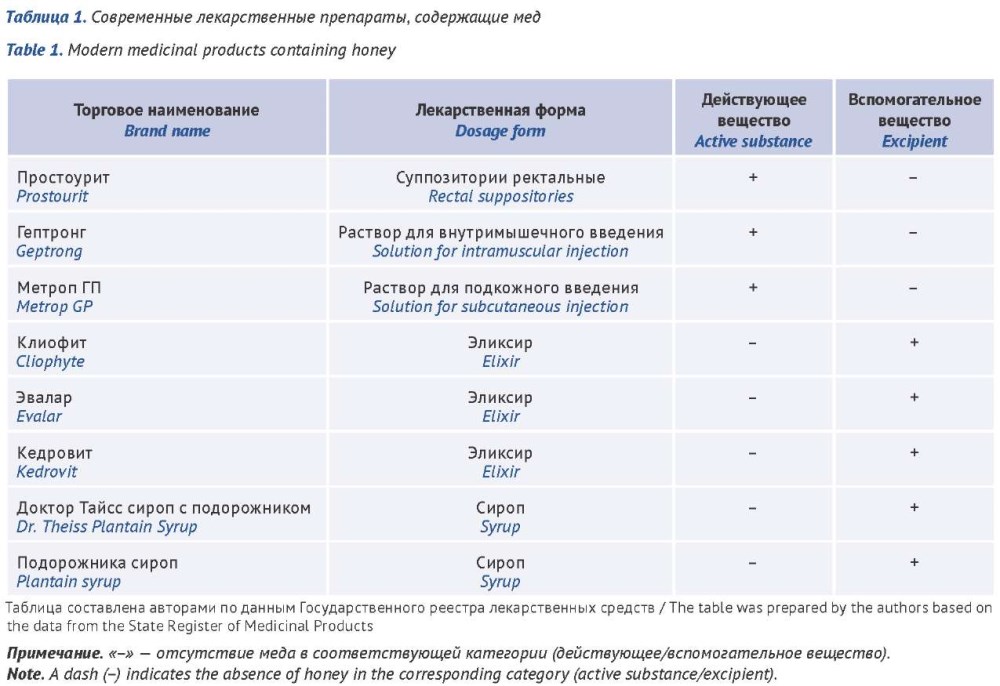
INTRODUCTION. The Russian Federation does not have a pharmacopeial standard for honey as a pharmaceutical substance, and the current regulatory documents (GOST 19792-2017, foreign pharmacopoeias) impose different quality requirements that vary in terms of controlled parameters, test methods, and acceptance criteria. A comparative analysis of national and foreign requirements for honey quality is therefore relevant for the development of unified approaches to pharmacopeial standardization.
AIM. To perform a comparative analysis of the honey quality parameters specified in foreign pharmacopoeias and GOST 19792-2017 in order to substantiate a list of critical quality attributes that can be used in the development of a pharmacopeial monograph for honey as a pharmaceutical substance.
DISCUSSION. A literature review was conducted using the PubMed, Google Scholar, and eLIBRARY.RU databases for the period 2015–2025. A comparative analysis of the current monographs on honey in the European Pharmacopoeia (Ph. Eur.), the Korean Pharmacopoeia (KP), the Pharmacopoeia of the People’s Republic of China (ChP), the Japanese Pharmacopoeia (JP), and the United States Pharmacopeia (USP), as well as GOST 19792-2017 (GOST), was performed. Eight medicinal products (MPs) containing honey are registered in the Russian Federation; of these, honey serves as the active substance in three products and as an excipient in five. The comparative analysis of the monographs revealed substantial differences in the approaches to the standardization of honey as a pharmaceutical substance. In the Ph. Eur., identity is confirmed by the sugar profile using thin-layer chromatography (TLC); in the USP, by a qualitative reaction for proline. The content of glucose, fructose, and their ratio is determined only in the ChP and GOST. The most critical discrepancies were found for the parameters related to the thermal treatment of honey and its adulteration: the limits for 5-hydroxymethylfurfural (5-HMF) range from 25 million–1 in GOST to 80 ppm in the Ph. Eur. and the KP, while the lists of controlled impurities in the ChP, the JP, and GOST do not coincide. Control of impurities indicative of adulteration is provided for in the ChP, the JP, and GOST, but the criteria differ: the qualitative reaction with iodine for starch and dextrin (the ChP, the KP, the JP), the reaction with tannic acid (the KP, the JP), TLC for oligosaccharides and HPLC for sucrose/maltose (the ChP), and the mass fraction of sucrose (GOST). Determination of the diastase number is provided for only in GOST. Foreign manufacturers of MPs rely on the requirements of the Ph. Eur., while domestic manufacturers rely on GOST and internal specifications, creating obstacles to harmonization. The data obtained substantiate the need for the unification of requirements for honey as a pharmaceutical substance and the revision of the current regulatory documents.
CONCLUSIONS. For the first time, a systematic comparative analysis of Russian and foreign regulatory requirements for honey quality was conducted. The analysis allowed us to identify critical discrepancies, establish a list of the most important honey quality parameters requiring harmonization, and develop a prospective list of quality parameters that can be used in the development of a pharmacopeial monograph for honey as a pharmaceutical substance.
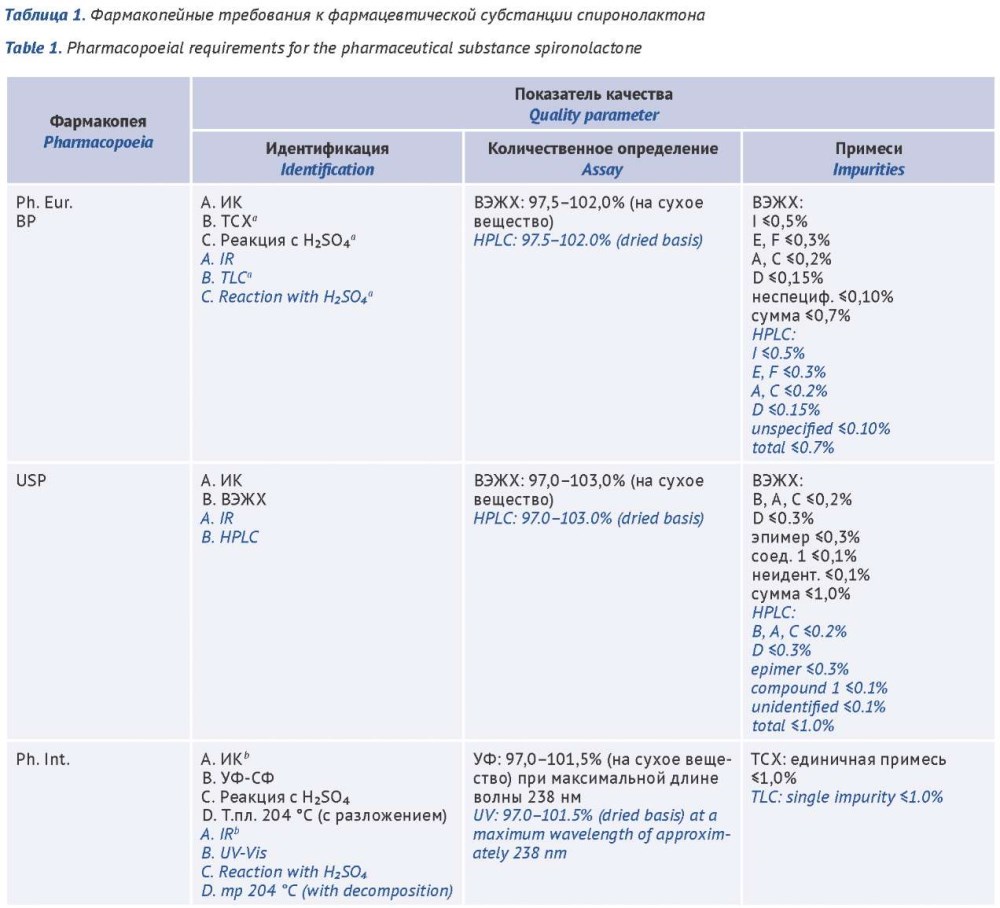
INTRODUCTION. Spironolactone, included in the list of vital and essential medicinal products, is used for chronic heart failure, arterial hypertension, and primary hyperaldosteronism, which imposes stringent quality requirements for its pharmaceutical substance and finished dosage forms. Current pharmacopoeial requirements for spironolactone differ significantly in impurity control, identification, and assay methods, creating regulatory barriers and complicating quality assessment of the products. To address this problem, the authors performed a comparative analysis of pharmacopoeial requirements to identify areas for harmonization.
AIM. This study aimed at a comparative evaluation of the requirements of foreign pharmacopoeias and the State Pharmacopoeia of the Russian Federation for the quality of the pharmaceutical substance and tablet dosage forms of spironolactone, in order to identify directions for harmonization of national pharmacopoeial monographs.
DISCUSSION. The objects of analysis were the current monographs of the European Pharmacopoeia (Ph. Eur.), the United States Pharmacopeia (USP), the British Pharmacopoeia (BP), the Indian Pharmacopoeia (IP), the Japanese Pharmacopoeia (JP), the Chinese Pharmacopoeia (ChP), the International Pharmacopoeia (Ph. Int.), the Korean Pharmacopoeia (KP), and the State Pharmacopoeia of the Russian Federation (SP RF). The methodology was based on a comparative analysis of the monographs for key quality attributes: Identification, Assay, Related substances, and Dissolution (for tablets). The comparative analysis showed that the most stringent and detailed requirements for related substances are imposed by Ph. Eur. and USP, where the determination is performed by HPLC. Several other pharmacopoeias (SP RF, IP, ChP) still use less selective methods (UV spectrophotometry, thin-layer chromatography) and general acceptance criteria for impurities, which do not allow full control of the stability and purity of the substance. For finished dosage forms, a high degree of harmonization of the dissolution test conditions was found, while differences remain in acceptance criteria and analytical methods for quantitation. A priority area for the development of national pharmacopoeial monographs is the implementation of chromatographic methods and the harmonization of impurity limits with leading international standards.
CONCLUSIONS. The identified differences in pharmacopoeial approaches confirm the advisability of revising national standards toward harmonization with the requirements of Ph. Eur. and USP. Incorporation of a highly specific HPLC method and detailed impurity specification into the SP RF pharmacopoeial monographs for spironolactone‑based medicinal products will help ensure the quality and safety of these products.
INTRODUCTION. The relevance of this study is driven by the growing interest in polypore fungi as an accessible and renewable source of biologically active compounds with proven pharmacological activity, particularly phenolic and triterpene compounds. Despite the widespread occurrence of fomitoid polypores, their raw materials remain largely unstandardized and are not represented in the pharmacopoeias of most countries, which limits their introduction into medical practice. The lack of unified approaches to quality assessment and the selection of analytical markers, combined with the high variability of their chemical composition, complicates the standardization of the raw material. Therefore, substantiating quality criteria and standardization methods for raw materials derived from fomitoid polypores is an important task.
AIM. To substantiate quality criteria for raw materials of fomitoid polypores that can be used for the development of regulatory documentation on the quality of fungal raw materials.
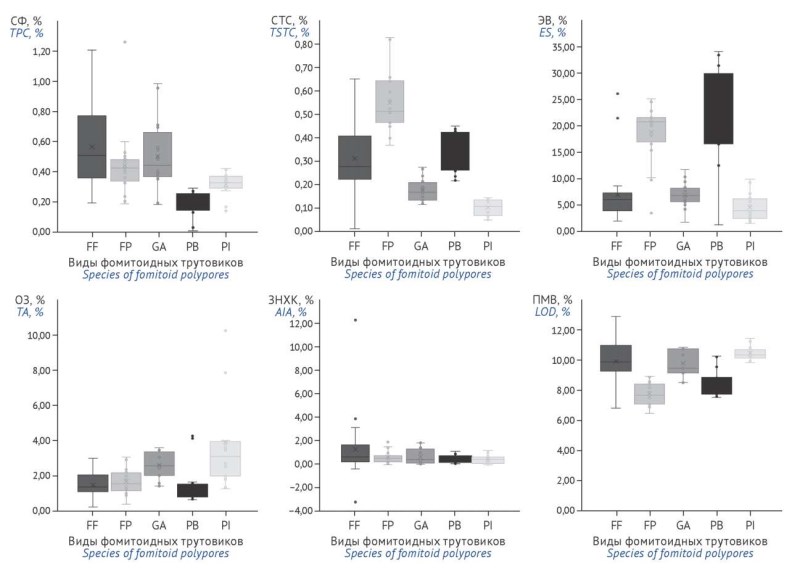
MATERIALS AND METHODS. At least 20 batches of fruiting bodies of Fomes fomentarius (L.) Fr., Fomitopsis pinicola (Sw.) P. Karst., Ganoderma applanatum (Pers.) Pat., Piptoporus betulinus (Bull.) P. Karst., Phellinus igniarius (L.) Quél., collected in various regions of the Republic of Belarus and the Russian Federation, were studied. The following parameters were determined using spectrophotometric and gravimetric methods: loss on drying, total ash, acid-insoluble ash, total extractives, total phenolic compounds, and total steroid and triterpene compounds.
RESULTS. Inter- and intraspecific variability of quality indicators was demonstrated. It was established that, depending on the species, the specification limits for total phenolic compounds range from 0.10% to 0.25%, for total steroid and triterpene compounds from 0.05% to 0.30%, and for extractives from 0.5% to 14.0%. The values of total ash, acid-insoluble ash, and loss on drying fall within the ranges of 1.0–5.0%, 1.0–3.0%, and 10.0–12.0%, respectively.
CONCLUSIONS. Quality indicators for raw materials of fomitoid polypores have been developed and may be used for the preparation of regulatory documentation on the quality of raw materials of fungal origin.
INTRODUCTION. Trace elements can influence the pharmacological effect of herbal medicinal products. However, their quality control is currently limited to assessing the content of only toxic elements (Pb, Cd, Hg, As), without evaluating the complete trace element profile. Therefore, studies on the elemental composition of complex herbal products available on the pharmaceutical market are highly relevant.
AIM. To perform a comparative assessment of the trace element content (Al, Ba, Cu, Fe, Mn, Sr, Zn) in pharmacopoeial pectoral species.
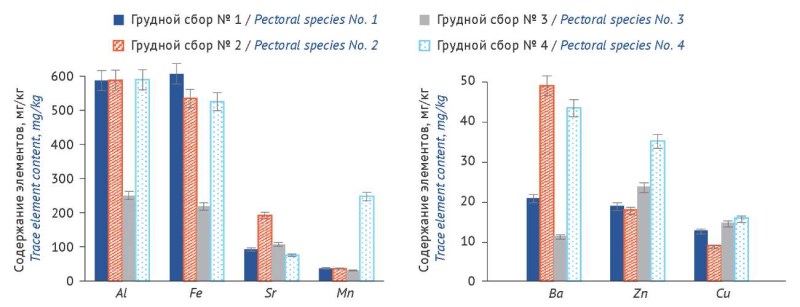
MATERIALS AND METHODS. The objects of the study were samples of pectoral species No. 1, No. 2, No. 3, and No. 4 produced by Russian manufacturers. The content of the seven elements specified in the aim was determined using inductively coupled plasma atomic emission spectrometry (ICP-AES).
RESULTS. The content (mg/kg) of trace elements in the pectoral species samples varied within the following ranges: Fe 134.9–707.8; Mn 25.6–248.7; Cu 3.8–16.9; Zn 4.3–35.7; Al 184.2–769.8; Ba 8.4–43.8; Sr 74.3–254.6. The elements differed in concentration levels: Fe and Al — high; Sr and Mn — medium; Ba, Cu, and Zn — low. The estimated daily intake for humans was: Ba 0.08–0.43 mg/day; Cu 0.03–0.16 mg/day, which is 3–18 and 19–100 times lower, respectively, than the established acceptable daily intake levels (1.4 and 3.0 mg/day) set by Decision No. 138 of the Board of the Eurasian Economic Commission (EAEC).
CONCLUSIONS. For the first time, a comparative analysis of the trace element content (Al, Ba, Cu, Fe, Mn, Sr, Zn) in pectoral species No. 1–4 from different manufacturers has been conducted. The trace element profile revealed that the content of trace elements in individual mixtures varied by a factor of 3–8, depending on the sources of raw materials selected by the manufacturer. Distinct accumulation patterns were identified for each mixture: pectoral species No. 1 showed the highest Fe content; No. 2 — the highest Sr content; and No. 4 — the highest levels of Mn, Cu, and Zn. These findings should be taken into account when using phytotherapy for the treatment and prevention of upper respiratory tract diseases.

ISSN 3034-3453 (Online)





























