


MAIN TOPIC: LIFE CYCLE OF A MEDICINE

The State Pharmacopoeia is a strategic document that stipulates safety and quality standards in Russia. Its current progress directly correlates with the functioning of Scientific Centre for Expert Evaluation of Medicinal Products (Ministry of Health of the Russian Federation). Valeria L. Bagirova, Dr.Sci. (Pharm.), Professor, Director of Institute of Pharmacopoeia and Medicinal Product Standardisation, follows the key milestones in her exclusive interview. It places special emphasis on harmonisation of Russian provisions with the EAEU Pharmacopoeia and the leading world pharmacopoeias. Active international cooperation and over 45% of new monographs in edition XV confirm the dynamic development of the pharmacopoeia. The interview demonstrates the creation of a common pharmacopoeial area that would provide the patients with high-quality and safe medicinal products.
PREPARING REGISTRATION DOCUMENTS
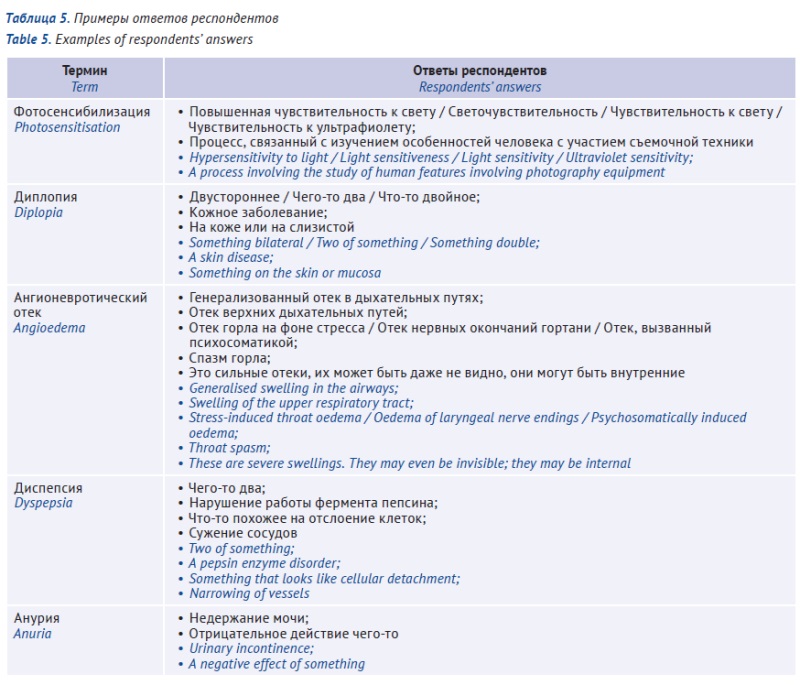
INTRODUCTION. The information that is intended for a patient and included in the package leaflet for a medicinal product should ensure the effective and safe use of the medicinal product through appropriate explanations. However, the appropriate level of detail and the necessity of defining each medical term are not clear.
AIM. This study aimed to test how respondents without medical education, potentially representative of the general patient population, understand medical terms randomly selected from approved package leaflets for medicinal products authorised in the Russian Federation, as well as to conduct a statistical analysis of the relationships between the respondents’ demographics and test results.
MATERIALS AND METHODS. A Google Forms-based comprehension test evaluated the understanding of 11 random terms from approved package leaflets for medicinal products authorised in the Russian Federation. The statistical analysis of comprehension included data from 63 respondents. The study collected the respondents’ demographic characteristics, including their level of education and age.
RESULTS. The respondents overestimated their comprehension of medical terms, giving incorrect definitions to some terms that they previously deemed to be comprehensible. Overall, the respondents provided correct definitions for less than 80% of the terms tested. The most comprehensible terms were “rhinitis” (66.07%), “angioedema” (28.57%), “exanthema” (25.00%), and “anuria” (25.00%). Those who had a higher education or worked with documents were more likely to define the terms correctly.
CONCLUSIONS. Package leaflets containing medical terms, regardless of how frequently these terms are used outside of healthcare settings, should be designed taking into account the heterogeneity of the target audience due to, among other things, differences in education. The terms that cause the greatest difficulty in user testing should be further explained or replaced with more understandable synonyms.
PRECLINICAL AND CLINICAL STUDIES
INTRODUCTION. The administration of 5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (TFISA) into the eyes can reduce intraocular pressure. Currently, this novel selective carbonic anhydrase II inhibitor is at the preclinical research stage. The excretion of TFISA and its metabolites has not been studied yet.
AIM. This study aimed to calculate the parameters of urinary and faecal excretion of TFISA and its major metabolites in rats.
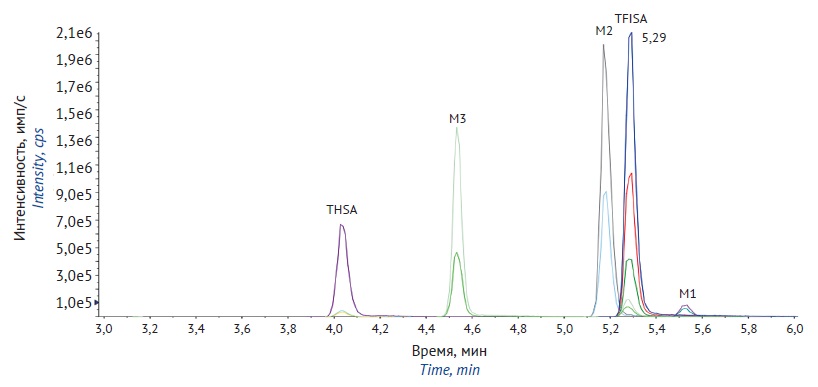
MATERIALS AND METHODS. The study was performed in 6 Wistar rats (3 males and 3 females). The rats received an instillation of 1% TFISA ophthalmic suspension in each eye at a dose of 3.7 mg/kg. Stool samples were taken prior to TFISA administration and at multiple intervals up to 360 h afterwards. Excreta were collected using metabolic cages. The study used high-performance liquid chromatography with tandem mass spectrometry (HPLC–MS/MS) to measure the concentrations of TFISA, N-acetyl-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (M2), and N-hydroxy-5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonamide (M1) with its degradation product 5-[5-(trifluoromethyl)-1,2-oxazole-3-yl]-furan-2-sulfonic acid (M3) in the excreta.
RESULTS. The study involved full validation of bioanalytical procedures for the determination of TFISA and its metabolites in rat excreta. The analytical ranges were 10–10,000 ng/mL for the urinary levels of TFISA, M2, and M3; 1–1,000 ng/mL for the urinary levels of M1; 10–4,000 ng/g for the faecal levels of TFISA; 5–2,000 ng/g for the faecal levels of M2 and M3; and 1–1000 ng/g for the faecal levels of M1. Out of the total amount eliminated, 45.7±2.0% of unchanged TFISA, 38.7±2.7% of TFISA in the form of M1 and its degradation product M3, and 4.0±0.6% of TFISA in the form of M2 were excreted in urine, while 8.2±1.0% of unchanged TFISA and 3.3±0.2% of the N-hydroxy metabolite M3 were excreted in faeces. TFISA elimination continued for up to 336 h after administration.
CONCLUSION. The developed and successfully validated bioanalytical procedures were used to study the excretion of TFISA and its metabolites in rats after single-dose administration of the ophthalmic TFISA suspension. According to the results, TFISA is predominantly excreted unchanged in urine. In addition, TFISA is eliminated as its N-hydroxy metabolite, which almost completely degrades to the sulfonic acid derivative during the sampling process. The N-acetyl metabolite of TFISA is minor and is exclusively excreted in urine.
INTRODUCTION. Colon cleansing plays an essential role in preparation for diagnostic procedures and surgical interventions. One of the key factors ensuring high-quality colon cleansing is the correct choice of a medicinal product that affects the intestinal contents and removes them from the body. Planning and conducting clinical trials for new effective colon cleansing medicines that are well-tolerated and have a favourable safety profile is an urgent task for health care. Currently, there are no regulatory documents on planning a clinical trial programme for colon cleansing medicinal products in the Russian Federation or the Eurasian Economic Union.
AIM. This study aimed to prepare recommendations for planning a clinical trial programme for bowel cleansing medicinal products.
DISCUSSION. Main provisions of the Guidelines of European Medicines Agency (EMA) were analysed for new medicinal products used in chronic constipation. The Guidelines define specific conditions for pharmacological studies and confirmatory clinical trials (regarding choice of design, study population, criteria for assessing efficacy and safety), as well as specific requirements for clinical trials in paediatric and elderly patients. Particular features of clinical trials for bowel cleansing medicines include patients with no comorbidities other than bowel disease; confirmatory clinical trials designed as randomised, active-control studies in parallel groups; safety assessment for a single-use medicinal product; long-term safety studies not required.
CONCLUSIONS. The main research models described in EMA Guidelines on clinical trials of bowel cleansing medicinal products enable a reliable assessment of safety and efficacy of the above medicines and can serve to prepare guidelines on clinical trial planning for bowel cleansing medicinal products.
PHARMACEUTICAL MANUFACTURING
INTRODUCTION. Ethylthiobenzimidazole fumarate (ETBIF) is a new actoprotector and adaptogenic active pharmaceutical ingredient (API). ETBIF was found to be effective given in short intervals; side effects related to ETBIF stomach solubility are known. Therefore, it is essential to develop a matrix dosage form with a specific release profile that can neutralize these side effects, prolong dosing intervals, and increase treatment compliance.
AIM. This study aimed to develop composition and production technology of ETBIF matrix tablets in accordance with a specified release profile (total release within 12 hours; not more than 50% of ETBIF released in acidic environment within the first two hours; the rest evenly released over the next 10 hours in the intestine-simulating environment).
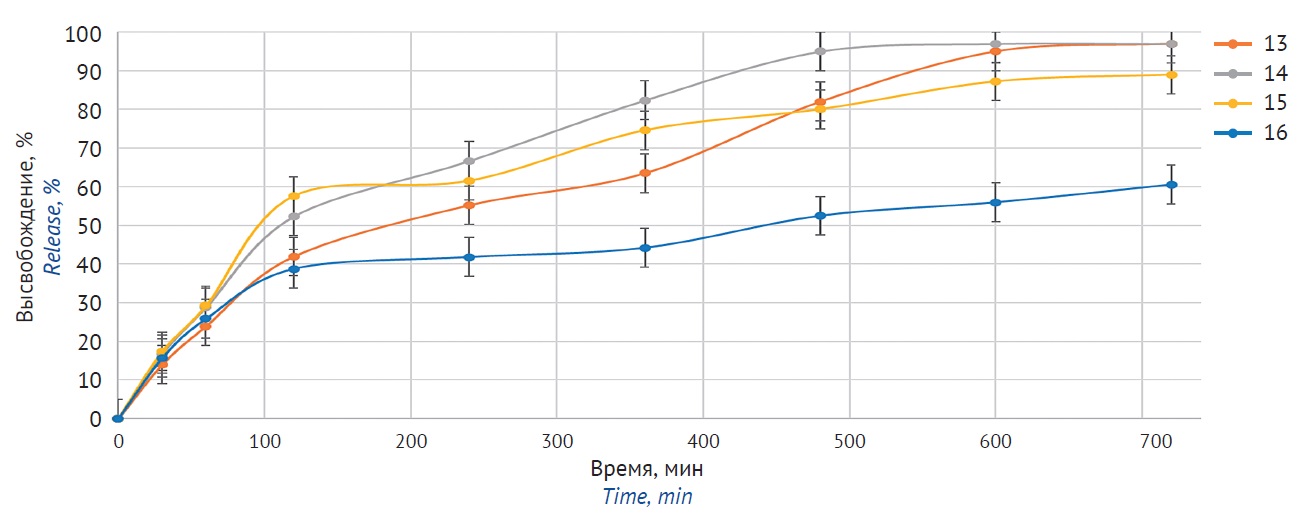
MATERIALS AND METHODS. The study examined ETBIF (API) as well as hydroxypropylmethylcellulose (hypromellose) and ethyl cellulose-based matrix ETBIF tablets. Mechanical properties of the tablets were examined, in particular hardness and friability. ETBIF release profiles were studied in different phosphate buffer solutions (pH 1.2, 4.5, and 6.8) using a dissolution tester and UV spectrometry.
RESULTS. The authors studied ETBIF release kinetics from matrix systems with various matrix-forming components of different origin. Hypromellose-based compounds enabled prolonged ETBIF release (90% after 12 h). Ethyl cellulose compounds showed 60 to 90% release, with API mostly releasing in acidic environment. Adding an acid does not affect ETBIF release from matrix tablets.
CONCLUSIONS. A composition of matrix tablets has been developed that meets the specified release profile. Hypromellose-based tablets showed the most complete and uniform release of the substance in an environment simulating gastrointestinal tract. Adding an acid does not affect ETBIF release from matrix tablets, despite its increased solubility in the acidic environment.
INTRODUCTION. Pre-treatment of local herbal substances is an essential growth vector in pharmaceutical industry. Canada goldenrod (Solidago canadensis L.) is a promising source of anti-inflammatory and diuretic substance widespread in Russia and Belarus. Pre-treatment of herbal substances increases the yield of biologically active substances (in particular flavonoids) during extraction, a trait useful for obtaining tinctures and extracts of Canada goldenrod herbs.
AIM. This study aimed to develop processing technology of tinctures and extracts obtained from pre-treated Canada goldenrod allowing to increase the content of flavonoids.
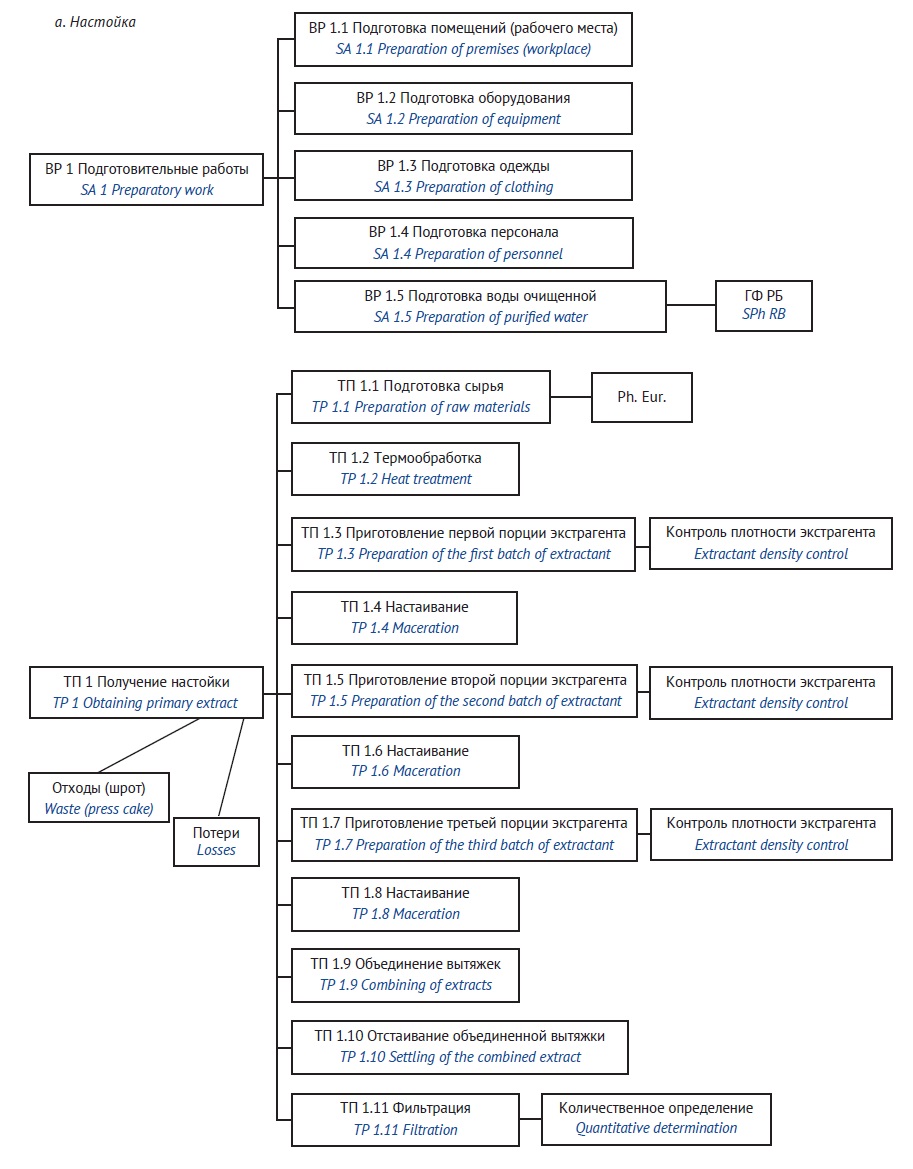
MATERIALS AND METHODS. Canada goldenrod herb was the study object. Four pre-treatment options were studied: heat pre-treatment, defatting, and their combinations. The content of flavonoids was determined by high-performance liquid chromatography. Gas chromatography was used to define residual organic solvents.
RESULTS. The highest yield of flavonoids in tinctures was observed with ethanol volume fraction of 60–70%, raw materials to extractant ratio 1 g to 25 ml, grinding degree of raw materials 2,000 μm, and a settling time of the primary extract no more than four days for remaceration. The highest content of flavonoids in dry extracts is achieved with 90% relative distillation volume, 80 °C distillation temperature, 40 min minimum distillation time, 6 cm thickness of the distilled layer, and no more than 4 days settling time of the primary extract. The highest yield of flavonoids in the tincture is observed in heat pre-treatment of Canada goldenrod herb and in pre-treatment defatting of the herbal raw material for the dry extract.
CONCLUSIONS. Optimal technological parameters for production of Canada goldenrod herb tinctures and extracts have been established. The above technologies developed considering pre-treatment stage can be used to produce the specified extracts of Canada goldenrod herb enriched with flavonoids.
DEVELOPMENT AND VALIDATION OF RESEARCH METHODS
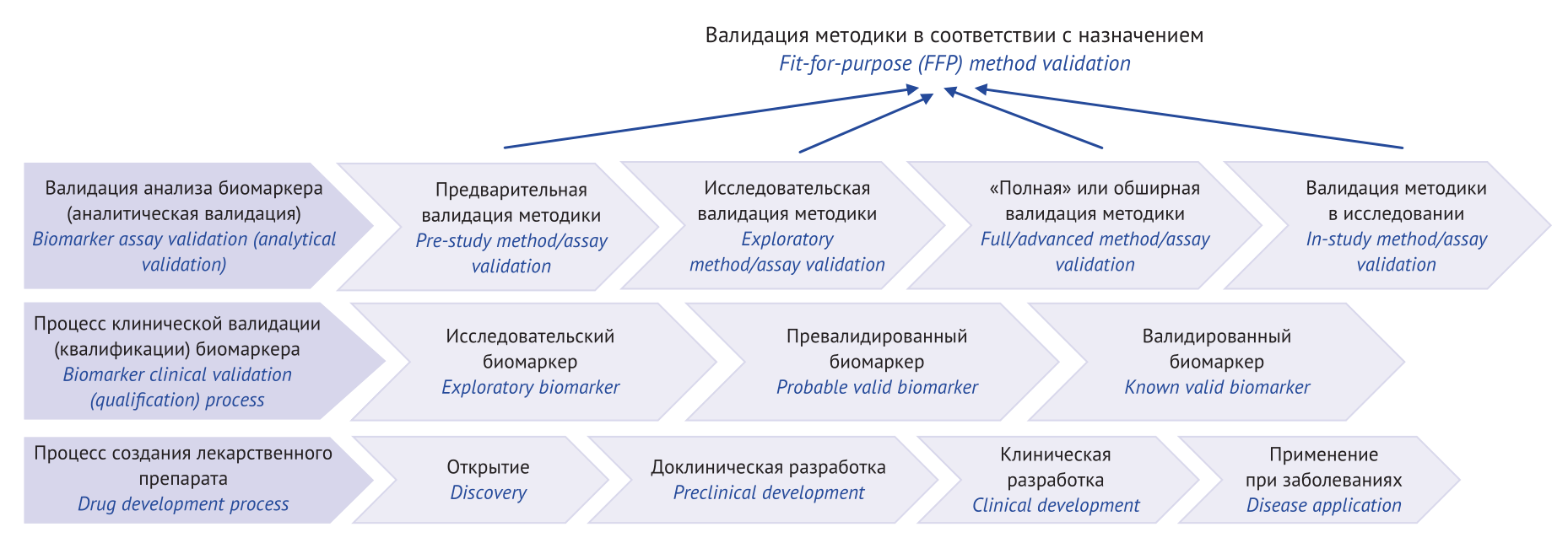
INTRODUCTION. Biomarkers are used to assess normal physiological processes in the body; to diagnose and select therapy for various diseases; and to develop new drugs. The increasing use of biomarkers as drug development tools necessitates improvements in analytical quantification methods.
AIM. This study aimed to review and summarise data on validation of bioanalytical methods for biomarker quantification.
DISCUSSION. The analysed regulatory documents were issued by International Council for Harmonisation (ICH), as well as Food and Drug Administration (FDA), including the Biomarker Assay Collaborative Evidential Considerations Writing Group and Critical Path Institute (C-Path); European Medical Agency (EMA); Eurasian Economic Commission (EEC); and other publicly available research platforms (including PubMed, Web of Science, RSCI (eLIBRARY.RU), and Google Scholar online databases) primarily published in 2014–2024. Analytical and clinical biomarker validation was examined, alongside with analytical validation stages and key validation parameters of the bioanalytical method depending on the objective (analysis for pharmacokinetics, bioequivalence, and toxicokinetics studies; biomarker analysis in drug development and for diagnosis in preclinical and clinical studies). Proposed is an algorithm for choosing biomarker analytical validation level depending on the method (chromatography, ligand-binding methods using reagent kits for various purposes) and the issues to be addressed.
CONCLUSIONS. Confirming biomarker method suitability as per the objectives is similar to a common validation procedure of bioanalytical methods. A broad and detailed scientific discussion of biomarker analysis validation is needed, since a comprehensive scheme developed for this complex procedure will contribute to more reliable use of biomarkers, improved quality of studies within this process, and resulting safe and effective new medicinal products.
INTRODUCTION. Red-stem buckwheat (Fagopyrum rubricaulis, Polygonaceae family) is characterised by a high content of flavonoids, primarily rutin. The aerial parts of the plant also contain quercetin, isoquercetin, orientin, vitexin, isoorientin, and other compounds. F. rubricaulis has antioxidant, cardioprotective, hypoglycaemic, and antibacterial pharmacological effects, which are attributed to the complex of flavonoids in the plant. Therefore, F. rubricaulis herb can be standardised using the content of total flavonoids. However, Russian publications lack information on the standardisation of F. rubricaulis herbal drugs. This motivates the development of an analytical procedure for the quantitative determination of flavonoids in F. rubricaulis.
AIM. This study aimed to develop and validate an analytical procedure for the quantitative determination of flavonoids in F. rubricaulis herb by spectrophotometry.
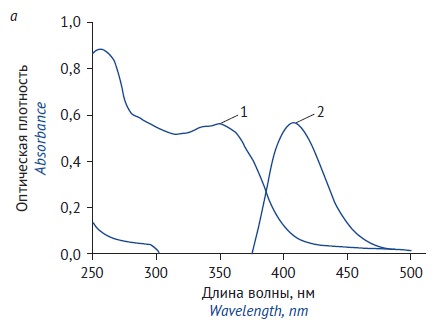
MATERIALS AND METHODS. The study involved extracting F. rubricaulis herb samples using several extraction solvents (purified water and 40%, 70%, and 95% ethanol). The presence of flavonoids in the herbal drug was confirmed by thin-layer chromatography (TLC). The content of flavonoids in F. rubricaulis herb was determined using a spectrophotometric procedure based on measuring the absorbance of the test solution in the presence of aluminium chloride. The analytical procedure was validated according to the State Pharmacopoeia of the Russian Federation (edition XV).
RESULTS. The TLC identification confirmed that the F. rubricaulis herb hydroalcoholic extracts contained flavonoids. The analytical procedure for the quantitative determination of flavonoids by differential spectrophotometry involved preliminary single-step hydroalcoholic extraction (herbal drug sample size: 1 g, accurately weighed; fragment size: ≤2 mm; extraction solvent: 40% ethanol; sample-to-solvent ratio: 1:25; extraction time: 30 minutes). The absorption maxima of the complexes of flavonoids with aluminium chloride were at 410 nm, which corresponded to the absorption maximum of the rutin standard solution. The content of total flavonoids was expressed as rutin. The analytical procedure was validated for specificity, linearity, repeatability, and intermediate precision. The developed analytical procedure was used to test 4 samples of F. rubricaulis herb manufactured by Parafarm LLC. The content of total flavonoids (expressed as rutin) ranged from 9.15±0.20% to 9.55±0.11%.
CONCLUSIONS. This study developed and validated a spectrophotometric analytical procedure for the quantitative determination of the content of total flavonoids (expressed as rutin), which can be used to standardise F. rubricaulis herbal drugs.
INTRODUCTION. Drugs based on rhizomes with roots of valerian (Valeriana officinalis L., family Valerianaceae) have sedative, antiarrhythmic, antispasmodic, anticonvulsant, anxiolytic effects and are widely used in psychoemotional and cognitive disorders, for the treatment of dysfunctions of the autonomic nervous system, including children, also for menopausal disorders. Biologically active compounds are contained not only in the underground, but also in the aboveground organs of the plant. This indicates the expediency of studying the qualitative chemical composition of valerian officinalis leaves to assess the possibility of use in medicine, subsequent standardization and the development of new phytopreparations based on them.
AIM. Comparative phytochemical analysis of biologically active substances of leaves and rhizomes with roots of Valeriana officinalis by thin-layer chromatography.
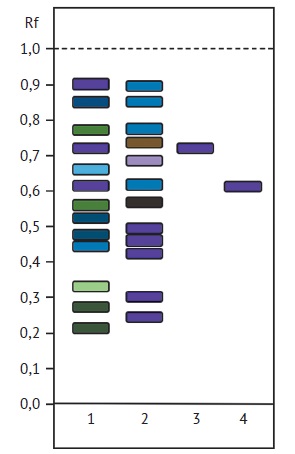
MATERIALS AND METHODS. Objects of research: leaves and rhizomes with roots of valerian harvested in various districts of the Minsk region of the Republic of Belarus in August 2023, dried by air-shade drying, ground to a coarse powder. The method of thin-layer chromatography on plates with a layer of silica gel (Sorbfil PTX-AF-A) was used to detect flavonoids and phenolic carboxylic acids. Chromatography was performed in the following mobile phases: ethyl acetate — formic acid anhydrous — water (8:1:1 and 10:2:3), acetic acid 2 and 15%; 0.020% solutions of rutin, quercetin, chlorogenic acid, and caffeic acid in 96% ethanol were used as comparison solutions. To detect sesquiterpene acids, chromatography was performed on plates with a layer of silica gel (Merck TLC Silica gel 60 F254) in the following mobile phases: glacial acetic acid — ethyl acetate — cyclohexane (2:38:60), ethyl acetate — hexane (10:90), acetone — hexane (1:2), hexane — ethyl acetate-glacial acetic acid (65:35:0.5). 0.025% solutions of valerenic acid and acetoxyvaleric acid in 96% ethanol were used as comparison solutions.
RESULTS. The conditions of thin-layer chromatography have been selected to separate and identify the chemical components of leaves of Valeriana officinalis, as well as a comparative analysis with biologically active substances of rhizomes with roots of V. officinalis. It was revealed that the leaves of V. officinalis contain flavonoids, tannins and saponins, rhizomes with roots contain phenolic compounds, mainly tannins. The best separation of flavonoids and phenolic carboxylic acids was achieved in the solvent system formic acid anhydrous — water — ethyl acetate (1:1:8). It was found that extracts from the leaves of V. officinalis contain rutin and chlorogenic acid, do not contain quercetin and caffeic acid. The optimal mobile phase for the separation of sesquiterpenic acids from valerian leaf is the glacial acetic acid — ethyl acetate — cyclohexane (2:38:60) system.
CONCLUSIONS. It has been established that alcoholic extracts of rhizomes with roots and leaves of valerian have a similar qualitative composition of flavonoids, phenolic carboxylic and sesquiterpenic acids. Rutin, chlorogenic, valerenic, and acetoxyvaleric acids were found in all samples. Thus, V. officinalis leaves can serve as a source of flavonoids, phenolic carboxylic and sesquiterpenic acids.
QUALITY CONTROL

INTRODUCTION. Sterility testing of advanced therapy medicinal products (ATMP) implies several challenges, including limited volume of available sample, product sensitivity to test conditions, and short shelf life. Classical pharmacopoeial test methods often fail to confirm sterility in a timely and reliable manner. The resulting drug availability for patients is reduced, as well as the opportunity for fast treatment with high-quality medicines. Faster alternative microbiological methods that reflect ATMP specificity of advanced therapy medicinal products can speed up and enhance sterility control.
AIM. This study aimed to optimise ATMP sterility control using colorimetric carbon dioxide detection.
MATERIALS AND METHODS. We used an experimental sample of an ATMP as the test object. We challenged the system with the following test strains: Bacillus subtilis ATCC 6633, Pseudomonas aeruginosa ATCC 9027, Staphylococcus aureus ATCC 6538, Clostridium sporogenes ATCC 19404, Cutibacterium acnes (Propionibacterium acnes) NCTC 737, Candida albicans ATCC 10231, Aspergillus brasiliensis ATCC 16404, Aspergillus fumigatus VKPM F-62, Aspergillus terreus VKPM F-1269, Penicillium chrysogenum VKPM F-3. We used SA, SN, and iLYM bottles with pharmacopoeial growth media: modified tryptic soy broth for the BacT/ALERT 3D Dual T system, tryptic soy broth, and fluid thioglycollate medium in test tubes designed for sterility testing using direct inoculation. All manipulations took place in a Purifier Logic A2 laminar flow cabinet, with BacT/ALERT 3D Dual T system to automatically monitor cultures, detect contamination, and record microbial growth curves. We prepared artificially contaminated samples by adding 3 CFU/mL of each test strain. We inoculated 0.1, 0.5, and 1.0 mL sample volumes into appropriate media, performing 20 replicates for each condition. Growth media included both pharmacopoeial (tryptic soy broth, fluid thioglycollate) ones and modified media for BacT/ALERT (SA, SN, iLYM). We incubated the direct inoculation samples for 14 days. In the BacT/ALERT system, we ran a 7-day incubation with continuous monitoring at 32.5±2.5 °C for bacteria and 22.5±2.5 °C for fungi. We evaluated direct inoculation results visually and assessed BacT/ALERT results based on CO2 growth curves (alternative method).
RESULTS. The BacT/ALERT 3D Dual T system detected ≥80% of aerobic and anaerobic bacteria and ≥90 % of yeasts and molds at 0.5 mL sample volume using pre-aerated iLYM medium. In contrast, direct inoculation required 1.0 mL to reach similar detection levels. BacT/ALERT reduced the time to contamination detection by 0.38–2.4 days compared to direct inoculation, while identifying all test strains except Candida albicans that showed similar detectability in both methods. When using SA medium, false-negative results for molds made 13–70 % of cases, depending on the strain. Short-time iLYM aerating for 5–7 s before incubation reduced false negatives to 0–3 %. We successfully detected all tested microorganisms within 28 h, except for Cutibacterium acnes, which required up to 134 h.
CONCLUSIONS. The BacT/ALERT 3D Dual T system delivers higher sensitivity, reproducibility, and faster detection compared to traditional pharmacopoeial methods. False negative results for mold fungi in SA medium were as high as 70%. However, they can be significantly reduced using aerated iLYM medium. Both methods reliably detect all test organisms within 7 days, but alternative BacT/ALERT achieves detection 0.38–2.4 days sooner. Using a 0.5 mL sample volume with BacT/ALERT effectively detects contamination and reduces product use by half compared to pharmacopoeial testing (1.0 mL).
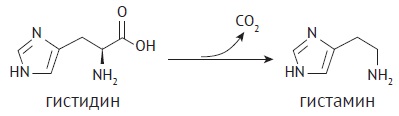
INTRODUCTION. Drug impurities lowering blood pressure can cause side effects in patients. These impurities include histamine and other depressor substances. The current General pharmacopoeial monograph “Test for Histamine” of the State Pharmacopoeia of the Russian Federation presents a quantification method of histamine impurity based on histamine interaction with H1 receptors of the guinea pig intestine. However, 3R concept (Replacement, Reduction, Refinement) introduced as an international standard and decision of leading pharmacopoeias to exclude in vivo histamine tests makes it necessary to develop in vitro methods for quantification of histamine impurity.
AIM. This study aimed to select an advanced in vitro method for quantification of histamine impurity as an alternative to in vivo tests.
DISCUSSION. Strategy of European Pharmacopoeia aimed at abandoning biological tests for histamine drug impurities was analysed. Scientific literature has shown the most common physicochemical and immunochemical methods for quantification of histamine impurity. The authors systematised test methods using high-performance liquid chromatography (HPLC). Indirect competitive heterogenous enzyme-linked immunosorbent assay (ELISA) was shown feasible for histamine quantification in the biological drugs. Choice between HPLC and ELISA was based the matrix of a test substance.
CONCLUSIONS. HPLC and ELISA are promising quantification methods of histamine impurity in biological products. In vitro methods are developed according to the composition, structure, and matrix properties of a test substance. Heterogeneous matrices, such as heparins, profit from HPLC, while ELISA is recommended for peptides and proteins, for example aprotinin.

ISSN 3034-3453 (Online)





























