




Содержание
Перейти к:
https://doi.org/10.30895/1991-2919-2026-16-2-163-178
Перейти к:
ВВЕДЕНИЕ. В соответствии с требованиями по процедуре регистрации лекарственных препаратов (ЛП) в регистрационное досье должны быть включены данные по доклиническому изучению фармакокинетики лекарственных средств. Однако вопрос объема предоставляемых данных для различных групп препаратов, а также определение того, какие фармакокинетические процессы и их параметры на каждом этапе жизненного цикла препарата должны быть изучены, остается открытым.
ЦЕЛЬ. Анализ данных литературы, отечественных и зарубежных методических документов по доклиническому изучению фармакокинетики лекарственных средств для выбора оптимальной стратегии сбора данных фармакокинетики на разных этапах жизненного цикла лекарственного препарата.
ОБСУЖДЕНИЕ. Материалы исследования — регуляторные документы, руководства по доклиническому изучению ЛП, научные статьи и иные литературные источники, находящиеся в открытом доступе (в том числе по данным электронных баз РИНЦ (eLIBRARY.RU), PubMed, Web of Science). Рассмотрены основные подходы к планированию исследований фармакокинетики для различных лекарственных препаратов (оригинальных, воспроизведенных, биологических и т.д.), включая выбор вида и количества лабораторных животных, исследуемых доз и временных точек отбора биоматериала.
ВЫВОДЫ. Исследования фармакокинетики лекарственных препаратов на этапе доклинических исследований необходимы для оптимизации структуры молекулы действующего вещества, выбора оптимального пути введения и оптимальной лекарственной формы, прогнозирования значений фармакокинетических параметров у человека, сокращения временных затрат и рисков при разработке безопасных и эффективных лекарственных средств. Предложены дизайны исследований фармакокинетики на этапе скрининга молекул, оптимизации молекул и выбора лекарственной формы для исследований различных групп препаратов.
Карлина М.В., Косман В.М., Макарова М.Н., Макаров В.Г. Подходы к дизайну исследования фармакокинетики лекарственных средств в доклинических исследованиях (обзор). Регуляторные исследования и экспертиза лекарственных средств. 2026;16(2):163-178. https://doi.org/10.30895/1991-2919-2026-16-2-163-178
Karlina M.V., Kosman V.M., Makarova M.N., Makarov V.G. Approaches to Drug Pharmacokinetics Study Design in Preclinical Trials (Review). Regulatory Research and Medicine Evaluation. 2026;16(2):163-178. (In Russ.) https://doi.org/10.30895/1991-2919-2026-16-2-163-178
Фармакокинетика (ФК) — раздел фармакологии, изучающий процессы абсорбции, распределения, метаболизма и выведения (adsorption, distribution, metabolism, excretion, ADME). В настоящее время исследования ФК являются ключевыми при разработке лекарственного препарата (ЛП). Как самостоятельный раздел фармакологии ФК была выделена в 1930-х гг., однако до 1960-х гг. рассматривалась исключительно в качестве академической дисциплины, не имеющей прикладного значения. Впервые практическая значимость ФК была показана при разработке математической модели оптимального режима применения сульфаниламидов, позволившей сократить количество нежелательных реакций [1].
Доклинические фармакокинетические данные должны быть включены в состав регистрационного досье на лекарственные препараты (модуль 4, «Доклинические исследования»)1. На основании подобных данных может быть проведен скрининг биологически активных молекул, оптимизация лекарственных форм (ЛФ), а также сделан прогноз потенциальных лекарственных взаимодействий [2]. Совокупность знаний о ФК, фармакодинамике (ФД) и токсичности позволяет выбрать ЛП с благоприятным фармакологическим профилем, определить оптимальные пути введения и схемы дозирования препаратов в клинической практике, а также дает понимание поведения действующего вещества (ДВ) в организме [3][4].
Дизайны ФК-эксперимента могут значительно различаться в зависимости от конечной цели исследователя: скрининговое исследование соединений, изучение ФК в ходе разработки ЛФ, изучение возможного фармакокинетического взаимодействия или регистрационные исследования ЛП. Для алгоритмизации возможных дизайнов исследования ФК необходимы сбор и систематизация имеющихся данных литературы.
Цель работы — анализ данных литературы, отечественных и зарубежных методических документов по доклиническому изучению фармакокинетики лекарственных средств для выбора оптимальной стратегии сбора данных фармакокинетики на разных этапах жизненного цикла ЛП.
При подготовке работы использовали регуляторные документы Международного совета по гармонизации (International Council for Harmonisation, ICH), Европейского агентства по лекарственным средствам (European Medicines Agency, EMA), Евразийской экономической комиссии (ЕЭК), а также находящиеся в открытом доступе научные статьи, преимущественно опубликованные в период 2014–2024 гг. Поиск производили в электронных базах данных PubMed, Web of Science, РИНЦ (eLIBRARY.RU) и поисковой системе Google Scholar. Ключевые слова, использованные в поисковых запросах: фармакокинетика, доклинические исследования, ADME, биоаналитические методы и их переводные аналоги.
Подробные рекомендации по дизайну и объему изучения ФК лекарственных средств (ЛС) на этапе доклинических исследований (ДКИ) в рамках документов Евразийского экономического союза (ЕАЭС) в настоящее время отсутствуют. В Решении2 в разделе 4.2.2 отмечено, что документы регистрационного досье по ФК-исследованиям включают результаты анализа всех процессов, происходящих с активным веществом и его метаболитами в живом организме, и охватывают изучение абсорбции, распределения, биотрансформации и выведения активного вещества и его метаболитов. Доклиническая оценка безопасности для регистрации ЛП должна включать доклинические ФК-исследования3. До начала клинических исследований (КИ) должны быть получены данные о системной экспозиции (токсикокинетике) на тех же самых видах животных, которые были использованы для изучения токсичности при многократном введении. Более подробные данные о ФК (включая сведения об абсорбции, распределении, метаболизме и выведении) у исследуемых видов животных должны быть доступны до назначения ЛП большому количеству субъектов или до начала III фазы КИ. Аналогичные требования содержит ICH M3 (R2)4: результаты исследований метаболизма и связывания с белками плазмы in vitro животных и людей следует оценивать до начала КИ. Сравнение метаболитов, образующихся у человека и животных, необходимо для оценки целесообразности проведения дополнительных испытаний, а также оценки вклада метаболита в общее действие ЛП: если метаболит характеризуется токсичностью и (или) его фармакологический эффект превышает 10%, то следует провести ФК-исследования этого метаболита. Такие исследования должны проводиться для поддержки КИ фазы III. Однако очевидно, что вклад метаболитов в фармакологическое действие необходимо оценить как можно раньше, т.к. метаболит может быть токсичным, обладать большим фармакологическим эффектом, что потребует его дополнительной характеристики и тестирования в ДКИ.
Результаты in vitro исследований связывания ДВ с белками, возможных путей метаболизма, проницаемости, а также оценка абсолютной биологической доступности (БД) вещества in vivo могут обосновать выбор ЛФ препарата и пути его введения, поэтому эти исследования необходимы уже на этапе фармацевтической разработки ЛП. Примером изучения ФК ДВ и оценки его абсолютной БД для обоснования разработки препарата для перорального введения является работа Г.Б. Колыванова и соавт. [5], в которой была оценена ФК кардиопротективного средства после однократного введения крысам внутривенно и внутрижелудочно. Установленная абсолютная БД позволила сделать вывод о возможности разработки ЛФ для приема внутрь.
В ICH S3B5 рассмотрены вопросы изучения распределения препаратов по органам и тканям; даны разъяснения, в каких случаях необходима оценка распределения при однократном и многократном введении ЛП. Оценку тканевого распределения и потенциала кумуляции большинства ЛП рекомендуется проводить после их однократного введения. Исследования при многократном введении оправданы в случае, если препарат характеризуется длительным периодом полувыведения, неполной элиминацией или имеет непредвиденную органную токсичность. В ICH S3B не конкретизируется, нужно ли проводить такие исследования активной фармацевтической субстанции (АФС) или ЛП и, в случае ЛП, на каком этапе разработки. Однако изучение процессов распределения необходимо для выявления органов и тканей с максимальным накоплением ДВ, детального изучения механизмов действия ЛС [6], а также создания систем адресной доставки ЛП или увеличения его биодоступности [7–9].
Наиболее подробно ДКИ ФК для различных групп препаратов (оригинальный препарат, воспроизведенный препарат, новая ЛФ препарата и др.) описаны в Руководстве по проведению доклинических исследований6, где выделена отдельная глава — «Методические рекомендации по проведению доклинических исследований фармакокинетики лекарственных средств». Однако в документе не указаны параметры оценки при скрининге новых молекул, перечень необходимых исследований АФС и ЛП на этапе разработки, а также объем исследования возможных ФК-взаимодействий.
В жизненном цикле ЛП можно выделить несколько этапов [10], часть которых подразумевает изучение доклинической ФК: синтез и скрининг молекул, оптимизацию структуры молекулы и технологии синтеза ДВ, доклиническую разработку ЛП (включая фармацевтическую разработку и ДКИ).
Поиск новых молекул начинается с выбора белка-мишени, вовлеченного в патогенез определенного заболевания человека. Затем проводится поиск соединения, которое может связываться с белком-мишенью. Для дальнейшей разработки молекула-кандидат должна обладать достаточной биологической активностью, профилем безопасности, должны быть предварительно получены фармакокинетические данные. Стратегия разработки ЛС включает такие подходы, как высокопроизводительный скрининг, секвенирование генома, использование микрочипов ДНК, робототехника, миниатюризация и методы биоинформатики [11]. Наряду с оценкой фармакологической активности и изучением токсических свойств новых молекул, ФК занимает одно из ключевых мест в скрининге, т.к. процессы всасывания, распределения, метаболизма и выведения напрямую определяют судьбу кандидатов в ЛС [12]. На этапе скрининга проводят исследования процессов ADME in vitro (метаболическая стабильность в микросомах печени животных и человека, метаболизм в системе цитохромов Р450, растворимость и проницаемость через монослой клеток Caco-2, связывание с белками плазмы крови), а также исследования этих процессов in vivo в совокупности в живом организме [13–15]. Однако необходимо отметить, что многие исследования in vitro достаточно длительны и являются дорогостоящими, кроме того на данном этапе количества образца ДВ для исследования, как правило, не так много. Учитывая вышесказанное, а также то, что этап скрининга преодолевает только незначительное количество кандидатов, рационально минимизировать объем исследований ФК. Поскольку основной целью изучения ФК при скрининге молекул является установление зависимости фармакологического эффекта от системной концентрации вещества [13] и наиболее важными параметрами ФК на этом этапе являются скорость выведения препарата и прогнозирование его клиренса (CL) [10][16], достаточно получить предварительную ФК с несколькими временными точками в предполагаемом интервале дозирования на одном виде животных.
Минимальный объем исследований на этапе скрининга (табл. 1) включает оценку параметров ФК при двух путях введения: внутривенном (обеспечивает абсолютную БД) и ином, установленном в исследованиях ФД и (или) планируемом в клинической практике. Представленный дизайн позволяет валидировать биоаналитическую методику для определения ДВ и (или) его метаболитов в плазме крови или обосновать необходимость модификации методики (например, для повышения чувствительности). В случае если известна биологическая мишень молекулы и ее локализация специфична, необходимо исследование распределения в органы и ткани [17]. Результаты этого исследования можно использовать для оптимизации химической структуры соединения и выбора ЛФ будущего ЛП для получения благоприятного фармакокинетического профиля. В ряде случаев альтернативой подобным экспериментам может выступать компьютерное моделирование [18].
Таблица 1. Возможный дизайн исследования фармакокинетики на начальных этапах изучения действующего вещества
Table 1. Approximate design of the pharmacokinetic study at the initial stages of active pharmaceutical substance study
|
Группа Group |
Доза и режим введения Dose and administration |
Вид, количество животных Species, number of animals |
Биоматериал, временные точки Biological sample, time points |
|
Этап скрининга молекул / Molecule screening stage |
|||
|
1 |
Субстанция ДВ, доза 1, однократное введение (доза и путь введения, установленные в исследованиях ФД) APS, 1 single, administration (dose and route established in PD studies) |
Крысы, 30 (3 особи на 1 временную точку) Rats, 30 (3 per time point) |
Отбор крови и при возможности органов-мишеней на 10 временных точках Blood sampling and, when possible, target organ sampling at 10 time points |
|
2 |
Субстанция ДВ, доза 1, однократное внутривенное введение (только при условии растворимости в воде) APS, 1 single, intravenous administration (only if soluble in water) |
Отбор крови на 10 временных точках Blood sampling at 10 time points |
|
|
Этап оптимизации молекулы / Molecule optimization stage |
|||
|
1 |
Субстанция ДВ, доза 1, однократное введение, неизмененная молекула (доза и путь введения, установленные в исследованиях ФД) APS, 1 single application of unchanged molecule (dose and route established in PD studies) |
Крысы, 30 (3 особи на 1 временную точку) или кролики (3 особи) Rats, 30 (3 per time point) or rabbits, 3 animals |
Отбор крови на 10 временных точках Blood sampling at 10 time points |
|
2 |
Субстанция ДВ, доза 1, однократное введение, оптимизированная молекула (доза и путь введения, установленные в исследованиях ФД) APS, 1 single application optimized molecule (dose and route established in PD studies) |
||
Таблица составлена авторами / The table was prepared by the authors
Примечание. ДВ — действующее вещество, ФД — фармакодинамика.
Note. APS, active pharmaceutical substance; PD, pharmacodynamics.
После выбора соединения-кандидата могут потребоваться исследования по оптимизации химической структуры (введение дополнительных функциональных групп, получение солей и т.д.), а также отработка технологии синтеза. На данном этапе оптимально параллельное проведение исследований ФК и ФД на одном виде животных для обеспечения прогностической ценности данных и построения релевантных ФД- и ФК-моделей [19]. Примером такого подхода может быть работа X. Zhang и соавт. [20], где была проведена оценка ФК и ФД субстанции рекомбинантного человеческого интерферона альфа-2b у китайских макак-резусов после изменения технологии ее очистки и продемонстрировано отсутствие значимого влияния изменений на изученные параметры.
При изменении химической структуры соединения ДВ необходимо дополнительное исследование ФД, ФК и токсичности модифицированной структуры. Оптимальным дизайном исследований по оценке модифицированной структуры может быть сравнительное исследование модифицированного и немодифицированного соединения-кандидата при однократном введении одним путем в одной дозе (табл. 1).
На этапе фармацевтической разработки и ДКИ изучение ФК позволяет оценить улучшение БД, разработать оптимальную ЛФ, обеспечить адресную доставку ДВ препарата, а также разработать воспроизведенный препарат с профилем безопасности и эффективности, сопоставимым с таковым у оригинального препарата. При выборе ЛФ препарата ведущей является биофармацевтическая концепция разработки ЛФ, основанная на создании ЛП с учетом фармацевтических факторов, таких как состав ЛП (в том числе вспомогательные вещества (ВВ)), вид ЛФ, путь введения, физико-химические свойства АФС, технологические условия производства [21], а также учитывающая результаты исследований, проведенных на этапе скрининга. При разработке ЛП в ЛФ для перорального приема безусловно важно понимание степени всасываемости вещества (его проникающей способности), стабильности в ЖКТ, что определяет выбор ВВ, также эта информация важна на этапе разработки воспроизведенных препаратов с точки зрения возможности проведения процедуры биовейвер.
Проведение экспериментов in vitro в ходе ДКИ регламентируется Решением Совета ЕЭК7. Примером использования информации о ФК для выбора оптимальной технологии и ЛФ для обоснования пути введения могут служить работы [22–25]. Так, в работе [22] авторы показали, что БД фтортиазинона из твердой дисперсионной формы при внутрижелудочном введении крысам гораздо выше по сравнению с кристаллической формой, что дало основание считать ее более подходящей для дальнейшей разработки лекарственной формы. Авторы работы [23] доказали, что БД куркумина при пероральном введении в виде микроэмульсии увеличивается, кроме того, при этом повышается проникновение его в головной мозг. В работе [24] показана перспективность использования нановезикулярных носителей для увеличения БД ропинирола. Авторы работы [25] определили абсолютную БД нового противопаркинсонического средства после внутрижелудочного введения мышам, которая составила 10–16%, что позволило говорить о потенциальной возможности разработки лекарственной формы для приема внутрь.
Для определения оптимального состава или пути введения будущего препарата на этапе фармацевтической разработки достаточно проведения сравнительного изучения ФК с определением концентрации ДВ только в крови (дизайн аналогичен исследованию оптимизированной молекулы, табл. 1), т.к. на данном этапе важно определить фармакокинетические преимущества/особенности того или иного состава или пути введения. При разработке сложных ЛФ необходимы более обширные исследования ФК, поскольку ЛФ могут менять не только БД, но и распределение ДВ в органы и ткани. Так, для липосомальных ЛФ, согласно Решению8, уже на этапе фармацевтической разработки рационально оценить и распределение в органы и ткани, что подтвердит ее успешность (в случае отрицательного результата потребуется доработка ЛФ). При разработке блок-сополимерных мицеллярных форм ФК может значительно меняться в зависимости от скорости CL мицелл и их диссоциации, а также скорости высвобождения ДВ из мицеллы. Аналогично меняется ФК ЛС в форме наночастиц, что учтено в Рекомендации9. Указано, что ФК подобных ЛП требует характеристики in vivo с оценкой ФК параметров как для общего, так и свободного ДВ в крови при разных дозах, определения путей метаболизма и оценки лекарственных взаимодействий. Также необходимо установить распределение в органы и ткани, значимые для клинического назначения, и пути введения препарата. Рекомендовано сравнивать ФК блок-сополимерного мицеллярного ЛП и его ДВ, вводимого в чистом виде, для подтверждения эффективности ЛФ. Для вводимых внутривенно препаратов такого рода может потребоваться оценка взаимодействия блок-сополимерных мицелл с белками и клетками крови, поскольку эти факторы способны влиять на распределение, стабильность и безопасность ЛС.
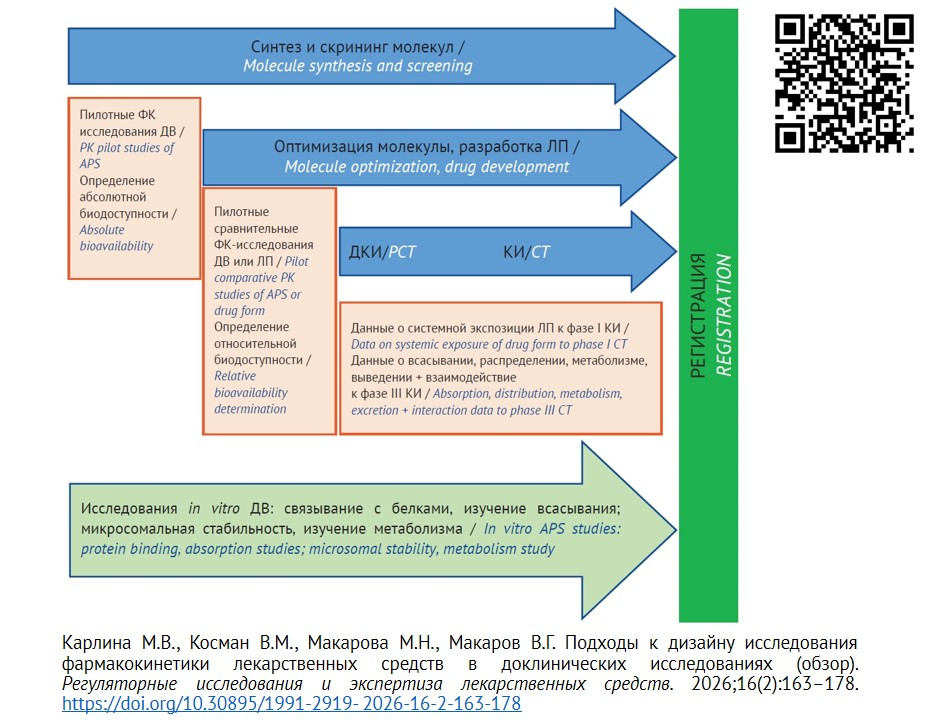
После выбора ЛФ следует провести комплекс исследований ФК, необходимых и достаточных для регистрации ЛП. Общая схема доклинического изучения ФК на предрегистрационном этапе приведена на рисунке 1. Объем исследований в зависимости от природы ДВ, его изученности и выбранной ЛФ может значимо отличаться.

Рисунок подготовлен авторами / The figure was prepared by the authors
Рис. 1. Доклиническое изучение фармакокинетики in vivo и in vitro на этапе фармацевтической разработки и предрегистрационном этапе
Примечание. ДВ — действующее вещество; ДКИ — доклинические исследования; КИ — клинические исследования; ЛП — лекарственный препарат; ФK — фармакокинетика.
Fig. 1. Preclinical in vivo and in vitro pharmacokinetics trials at the pharmaceutical development and pre-registration stages
Note. APS, active pharmaceutical substance; PCT, preclinical trial; СT, clinical trial; PK, pharmacokinetics.
Оригинальный ЛП. При регистрации препарата должны быть предоставлены данные о ФК, полученные как in vitro, так и in vivo.
В регистрационное досье на ЛП должны быть включены отчеты о следующих исследованиях in vitro: связывание ДВ с белками плазмы, метаболизм в печени и лекарственные взаимодействия ДВ10. Исследования in vitro целесообразно проводить с использованием тест-систем, полученных от человека, нежели тест-систем животного происхождения. Такие исследования могут быть выполнены параллельно с фармацевтической разработкой ЛП на субстанции ДВ.
Регистрационное досье на ЛП включает результаты исследований in vivo всех процессов, происходящих с ДВ и его метаболитами в живом организме. Программа исследований ФК должна обеспечивать возможность сравнения результатов, полученных в эксперименте на животных и человеке, а также экстраполяцию на человека результатов изучения ФК, полученных на животных моделях11.
Подробно объем и дизайн исследований фармакокинетики оригинального ЛС описан в Руководстве12 (рис. 2). Исследования ФК проводят, как правило, на здоровых животных одного пола. В ряде случаев существует необходимость изучать ФК и на животных с моделью патологического процесса (например, при изучении противоопухолевых ЛП).

Рисунок подготовлен авторами / The figure was prepared by the authors
Рис. 2. Возможный дизайн исследования фармакокинетики оригинального препарата
Fig. 2. Approximate design of pharmacokinetics study for original drug
В качестве экспериментальных моделей для исследований ФК как новых, так и уже известных ДВ чаще всего используют крыс [26][27], кроликов [28–30], реже используют животных других видов (собак [31], обезьян [32]). При проведении комплексных фармакологических исследований допускается использование мышей, морских свинок и кошек13. Выбор конкретных тест-систем (крысы, кролики, собаки, приматы) должен проводиться с учетом релевантности модели для конкретного ДВ [33], учитывать особенности метаболизма у разных видов животных [34] и отвечать требованиям дизайна планируемого исследования. Например, при исследовании высоких доз ЛП, необходимости введения ЛП полностью или получения большого количества биологических образцов в течение длительного периода времени требуется использование в эксперименте животного более крупного вида или более высокой организации [35].
Требования к количеству видов животных (не менее двух) при изучении оригинального препарата обоснованы необходимостью выявить возможные межвидовые различия в ФК препарата и наиболее точно экстраполировать значения ФК-параметров на человека за счет выявления общих закономерностей и различий в ФК веществ у экспериментальных животных [30][36].
Минимальное количество животных в группе, вводимых в эксперимент, — 5 (при реализации дизайна «одно животное — одна точка») или 6 (при условии отбора проб на всех временных точках от одной особи)14. В ряде исследований количество животных может быть увеличено [27][37], однако, как было показано при проведении ретроспективной оценки вариабельности параметров ФК [38], это не является оправданным, для изучения ФК на этапе ДКИ требования Руководства по проведению доклинических исследований лекарственных средств к количеству животных в группе являются достаточными.
Для внесения информации в регистрационное досье изучение ФК проводят на животных одного пола (как правило, самцах). Однако известно, что такие фундаментальные ФК-параметры, как клиренс (CL) и объем распределения (V), могут демонстрировать половые различия у самых разных видов животных: крыс, мышей, кроликов, хомяков и др. Например, различия в метаболизме лекарственных веществ у самцов и самок крыс часто являются результатом различий в экспрессии печеночных ферментов (цитохромы CYP2C11, CYP2C13 и CYP3A2 экспрессируются у самцов, тогда как CYP2C12 экспрессируется у самок) [39][40]. Необходимо отметить, что в ряде случаев установленные половые различия для животных не подтверждаются в клинике, однако фармакокинетические данные используют для оптимизации дозы в других доклинических исследованиях [41].
Для оригинальных ЛП необходимо изучение линейности ФК, т.к. это позволяет оценить предсказуемость изменения концентрации ДВ или его основного метаболита, обладающего фармакологическим действием, в крови в ответ на изменение вводимой дозы препарата [42–45]. Нелинейная ФК встречается достаточно часто и возникает, когда концентрации ДВ превышают возможности тех биологических объектов (ферментов, субстрата, рецепторов), которые отвечают за всасывание, распределение, метаболизм и выведение [1].
Изучение ФК при многократном введении ЛП позволяет прогнозировать возможную кумуляцию, изменение характера выведения и уровня стационарной концентрации ДВ в пределах интервала дозирования [46], а также оценить возможный вклад индукции метаболизма в изменение ФК [47].
Поскольку в состав ЛП помимо ДВ входят вспомогательные вещества, которые потенциально могут менять ФК ДВ, большую часть исследований корректнее выполнять непосредственно на ЛП. Если в состав ЛП введено ВВ, которое используют впервые, токсикологические и ФК исследования проводят в обязательном порядке15.
Воспроизведенный ЛП. В Решении Совета ЕЭК16 указаны требования к представлению информации в модуле 4 регистрационного досье на ЛП. В случае если воспроизведенный ЛП соответствует критериям, указанным в правилах проведения исследований биоэквивалентности ЛП, то необходимости в определении БД нет. В случае если ДВ воспроизведенного ЛП представляет собой производное ДВ ранее зарегистрированного ЛП, то можно представить литературное или экспериментальное подтверждение отсутствия изменений в ФК. Если таковые отсутствуют, то такое ДВ следует рассматривать как новое оригинальное ДВ.
Таким образом, необходимо литературное или экспериментальное подтверждение отсутствия изменений в ФК. При ДКИ воспроизведенного ЛП достаточно проведения сравнительного исследования с оригинальным ЛП на одном виде животных с анализом ДВ только в крови17. Дизайн такого исследования приведен в таблице 2.
Таблица 2. Возможный дизайн фармакокинетического исследования воспроизведенного препарата
Table 2. Approximate design of the pharmacokinetic study of generic drug
|
Группа Group |
Доза и режим введения Dose and administration |
Количество животных Number of animals |
Биоматериал, временные точки Biological sample, time points |
|
1 |
Доза 1, однократное введение воспроизведенного препарата Dose 1, generic drug single administration |
6 |
Отбор крови на 10 временных точках Blood sampling at 10 time points |
|
2 |
Доза 1, однократное введение оригинального препарата Dose 1, original drug single administration |
Таблица составлена авторами / The table was prepared by the authors
Гибридный ЛП. При разработке гибридного ЛП, как правило, происходят изменения ДВ, показаний к применению, дозировки, лекарственной формы или пути введения по сравнению с оригинальным препаратом.
Изменение ЛФ ЛП при сохранении аналогичного пути введения, а также изменение ЛФ и пути введения влечет за собой качественное и количественное изменение состава ВВ, что в ряде случаев может приводить к изменению ФД, ФК и профиля токсичности препарата. Классическим примером влияния замены ВВ является увеличение БД фенитоина при замене сульфата кальция на лактозу в капсулах, что вызвало вспышку отравления препаратом в 1968 г. в Австралии [48], более поздние исследования также показали несовместимость фенитоина с рядом ВВ, входящих в состав препарата [49].
В настоящее время взаимодействию действующих веществ и ВВ уделяют пристальное внимание [50][51], однако степень, в которой эти взаимодействия могут изменять биодоступность ДВ, не во всех случаях возможно спрогнозировать только по результатам испытаний in vitro (например, тест «Растворение»). В каждом конкретном случае это будет определяться такими факторами, как доза, терапевтический диапазон, место абсорбции, проницаемость и растворимость молекулы, а также метаболизмом, выведением, возможностью деградации в месте абсорбции [52]. Для новых ЛФ гибридных препаратов рекомендовано проведение изучения ФК/абсорбции, распределения, метаболизма и элиминации ДВ, поскольку при таких изменениях важно оценить форму фармакокинетической кривой и значение AUC18. Целью исследования новой ЛФ гибридного ЛП является оценка ее преимуществ, объем исследований аналогичен исследованиям воспроизведенного ЛП (табл. 2). Сравнивать новую ЛФ следует с уже существующей и оценивать относительную или абсолютную (при внутривенном пути введения) БД19. При изменении пути введения так же, как и в случае с новой ЛФ, рационально провести сравнительное изучение фармакокинетики. При изменении пути введения необходимо получать данные, характерные именно для нового пути введения: для офтальмологического пути введения — изучать распределение в тканях глаза; при создании препарата для введения в слуховой проход — изучать способность проникать через неповрежденную барабанную перепонку, для интраназального введения — оценивать проникновение в головной мозг, поскольку этот путь введения может обеспечить проникновение ДВ, минуя гематоэнцефалический барьер20.
Комбинированный ЛП. При использовании новых фиксированных комбинаций известных ДВ, которые уже были исследованы, информация о собственных фармакокинетических исследованиях может отсутствовать в регистрационном досье, если такое решение обосновано результатами исследований токсичности и экспериментальных терапевтических исследований21. В случае комбинации зарегистрированных ДВ, которая не была ранее одобрена регуляторными органами в качестве комбинированной терапии, следует провести ДКИ или КИ фармакокинетических взаимодействий, а для комбинации, основанием фармацевтической разработки которой является ФК-взаимодействие, последнее следует экспериментально подтвердить22. В случае если ДВ исследованы и охарактеризованы ранее, то регистрационное досье их новой фиксированной комбинации может не содержать результатов ДКИ и КИ по отдельности.
Документ23 оговаривает, что исследования ФК-взаимодействия в рамках ДКИ следует проводить на людях; ДКИ на животных могут быть проведены в случае, если нет значительных видовых различий, затрудняющих экстраполяцию результатов.
Таким образом, для комбинированных препаратов доклиническое изучение фармакокинетического взаимодействия в ряде случаев необходимо. Фармакокинетическое взаимодействие может проходить как на этапе всасывания, так и на этапе распределения, метаболизма и выведения [53], поэтому для некоторых комбинаций рационально определять ДВ не только в крови, но и в органах и тканях. Так, в работе [54] авторы при изучении комбинированного препарата Диоксазид показали, что комбинированное введение изониазида не влияет на БД диоксидина, однако наблюдали снижение концентрации изониазида в крови и увеличение его содержания в печени в сравнении с индивидуальным введением препарата в эквимолярных дозах. При оценке относительной БД и преимуществ комбинированного препарата могут быть использованы как субстанции ДВ комбинированного ЛП, так и зарегистрированные монопрепараты. Дизайн исследования ФК комбинированного ЛП приведен на рисунке 3.

Рисунок подготовлен авторами / The figure was prepared by the authors
Рис. 3. Возможный дизайн исследования фармакокинетики двухкомпонентного комбинированного препарата
Примечание. ДВ — действующее вещество; ЛП — лекарственный препарат.
Fig. 3. Approximate design of pharmacokinetics study, two-component combined drug
Note. APS, active pharmaceutical substance; PK, pharmacokinetic; D, drug.
Биологический ЛП. Перечень биологических ЛП включает широкий спектр препаратов различной природы, в связи с чем единой схемы проведения фармакокинетических исследований таких ЛП не существует. В соответствии с Решением24 информативными являются фармакокинетические исследования при однократном и многократном введении ЛП, токсикокинетические исследования, исследования распределения в тканях на релевантных видах животных. Оценка соотношения введенного и элиминированного ЛП не является необходимой частью исследования. Примерами изучения ФК биологических препаратов на этапе ДКИ могут служить работы [55–57].
В Решении отмечено, что при использовании белков с радиоактивной меткой важно доказать, что материал с радиометкой сохраняет активность и биологические свойства, эквивалентные таковым у немеченного вещества25. Получаемые данные может быть сложно интерпретировать из-за быстрого метаболизма белка in vivo или нестабильности связи радиоактивной метки с белком.
Значимой группой биопрепаратов являются ЛП на основе моноклональных антител, изучению которых посвящено сравнительно много публикаций. В отношении изучения их ФК авторы отмечают такие особенности, как возможности применения различных вариантов ВЭЖХ и, преимущественно, ИФА методов для анализа, а также длительная циркуляция этих молекул в организме [58–59]. Детализация объема ДКИ в отношении ФК в доступных работах не рассмотрена. Можно привести пример комплексного исследования, в котором ФК являлась лишь небольшой составной частью и его объем (внутривенное введение мышам в одной дозе, по 3 животных на временную точку) можно рассматривать только в аспекте первичной, предварительной оценки ФК нового лекарственного кандидата [60].
В случае генотерапевтических ЛП (ГТЛП) необходимо изучение биораспределения (персистенции26, CL и мобилизации27). При изучении персистенции допускается объединять исследования токсичности при однократном введении, фармакологической безопасности и исследования ФК28. При исследовании биораспределения рекомендуется29 провести анализ следующего набора органов и тканей / биологических жидкостей: головной и спинной мозг (шейный, грудной и поясничный отделы), печень, почки, легкие, сердце, селезенка, надпочечники, половые железы, ткани места введения и кровь. При отсутствии системного воздействия ГТЛП перечень исследуемых органов может быть сокращен [61]. В случае если в исследованиях специфической активности и токсичности используют различные виды лабораторных животных, то для оценки видовых различий и правильной экстраполяции результатов на человека биораспределение ГТЛП следует определять у каждого из видов, а также учитывать влияние модели заболевания. Продолжительность исследования биораспределения определяется как время до полного исчезновения обнаруживаемого сигнала или до достижения долгосрочного плато положительного сигнала. Следует помнить, что сигнал учитывают как в целевых, так и в нецелевых органах, а продолжительность обнаружения долгосрочного сигнала является предметом научного рассмотрения.
Стандартных исследований ФК ЛП на основе соматических клеток и препаратов тканевой инженерии не требуется. Однако если теоретическое обоснование в регистрационном досье на ЛП отсутствует, то необходимо изучить такие параметры, как жизнеспособность, долговечность, распределение, рост, дифференцировка и миграция клеток. В отношении соматотерапевтических ЛП и препаратов тканевой инженерии, синтезирующих активные биомолекулы, необходимо изучить распределение, продолжительность и объем экспрессии30.
Отметим, что в некоторых работах, посвященных созданию генотерапевтических и соматотерапевтических препаратов, рассматриваются различные аспекты этого процесса: вопросы качества, истории регистрации, основные направления клеточной терапии, анализируются данные об одобренных лекарственных препаратах клеточной терапии и тканевой инженерии, оцениваются проблемы и перспективы их использования, но такие работы не содержат детализации ДКИ таких препаратов в целом и ФК в рамках ДКИ в частности [62–65].
Биоаналогичный ЛП. Решение о необходимости проведения сравнительных исследований ФК биоаналогичных препаратов принимают в каждом отдельном случае исходя из данных, полученных in vitro. Наличие потенциально значимых количественных различий в показателях качества между рассматриваемым биоаналогичным (биоподобным) и оригинальным (референтным) ЛП, а также значимых различий в составе ЛП могут быть основанием для проведения ДКИ ФК при условии наличия релевантных животных для модели in vivo31.
В случае биоаналогичных (биоподобных) препаратов, как правило, проводят только одно исследование токсичности при многократном введении, включающее изучение токсикокинетики, определение титров антител, перекрестной реактивности и нейтрализующей способности. Указанные исследования позволяют определить значимые отличия токсических и (или) иммунных ответов между биоаналогичным и референтным препаратами32.
Примером сравнительных исследований биоаналогов могут служить работы [66][67].
Как регуляторные документы33, так и научные статьи [68][69] отмечают особенности и этапность исследований подобных биологических препаратов, отличающие их от исследований воспроизведенных препаратов химической природы. Решение о целесообразности проведения исследований in vivo принимают на основании результатов, полученных на предыдущих этапах разработки (аналитические сравнительные испытания, исследования in vitro), степени значимости выявленных различий и характеристики класса разрабатываемого продукта. Исследования in vivo не проводят, если отсутствует соответствующая биологическая модель. При разработке подобных биологических препаратов всегда требуется проведение клинических сравнительных испытаний, первым этапом которых являются сравнительные ФК/ФД-исследования, желательно в группе здоровых добровольцев, а при невозможности — в группе пациентов [70]. Отметим, что позиции доказательства биоподобия биопрепаратов только на основании сопоставления физико-химического и биологического сходства [71] можно противопоставить случай недостаточности такой характеристики (на примере эритропоэтина) и целесообразности проведения сравнительных исследований in vivo [72].
Проведен обзор данных литературы и нормативных документов, касающихся изучения фармакокинетики на доклиническом этапе разработки лекарственных препаратов. Согласно современным требованиям данные по фармакокинетике должны быть включены в состав регистрационного досье. Данные фармакокинетических исследований, полученные на разных этапах жизненного цикла лекарственного препарата, необходимы для оптимизации структуры молекулы, выбора оптимального пути введения и разработки оптимальной лекарственной формы, экстраполяции значений фармакокинетических параметров на человека, сокращения временных затрат и рисков при разработке безопасных и эффективных лекарственных средств.
Предложены дизайны исследований фармакокинетики на этапе скрининга молекул действующего вещества разрабатываемого лекарственного препарата, оптимизации молекул и выбора лекарственной формы для исследований различных групп препаратов.
Вклад авторов. Все авторы подтверждают соответствие своего авторства критериям ICMJE. Наибольший вклад распределен следующим образом: М.В. Карлина — идея публикации, обработка и систематизация данных, подготовка рукописи; В.М. Косман — обсуждение результатов, доработка текста; М.Н. Макарова, В.Г. Макаров — критический пересмотр текста публикации и иллюстративного материала.
Authors’ contributions. All the authors confirm that they meet the ICMJE criteria for authorship. The most significant contributions were as follows. Marina V. Karlina conceptualized the paper, processed and systematized the data, and prepared the manuscript. Vera M. Kosman discussed the results and revised the text. Marina N. Makarova, Valery G. Makarov critically revised the manuscript and illustrative material.
1. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
2. Там же.
3. Решение Коллегии ЕЭК от 26.11.2019 № 202 «Об утверждении Руководства по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов».
Миронов АН, ред. Руководство по экспертизе лекарственных средств. Т. 1. М.: Гриф и К; 2013.
4. ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. ICH; 2009. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-and-marketing-authorisation-pharmaceuticals-step-5_en.pdf
5. ICH S3B Pharmacokinetics: repeated dose tissue distribution studies. EMA; 1995. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s-3-b-pharmacokinetics-guidance-repeated-dose-tissue-distribution-studies-step-5_en.pdf
6. Миронов АН, ред. Руководство по проведению доклинических исследований лекарственных средств. Ч. 1. М.: Гриф и К; 2012.
7. Решение Совета ЕЭК от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
8. Решение Совета ЕЭК от 15.09.2020 № 111 «Об утверждении Руководства по фармакокинетическому и клиническому изучению биоэквивалентности липосомальных лекарственных препаратов для внутривенного введения».
9. Рекомендация Коллегии ЕЭК от 15.09.2020 № 15 «О руководствах по оценке качества и исследованию биоэквивалентности отдельных групп лекарственных препаратов».
10. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
11. Там же.
12. Миронов АН, ред. Руководство по проведению доклинических исследований лекарственных средств. Ч. 1. М.: Гриф и К; 2012.
13. Миронов АН, ред. Руководство по проведению доклинических исследований лекарственных средств. Ч. 1. М.: Гриф и К; 2012.
14. Там же.
15. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
16. Там же.
17. Миронов АН, ред. Руководство по проведению доклинических исследований лекарственных средств. Ч. 1. М.: Гриф и К; 2012.
18. Косенко ВВ, ред. Руководство по экспертизе лекарственных средств. Т. 1. Экспертиза отдельных групп лекарственных средств. М.: Типография «Миттель Пресс»; 2025.
19. Миронов АН, ред. Руководство по проведению доклинических исследований лекарственных средств. Ч. 1. М.: Гриф и К; 2012.
20. Косенко ВВ, ред. Руководство по экспертизе лекарственных средств. Т. 1. Экспертиза отдельных групп лекарственных средств. М.: Типография Миттель Пресс; 2025.
21. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
22. Рекомендация Коллегии ЕЭК от 02.09.2019 № 25 «О Руководстве по доклинической и клинической разработке комбинированных лекарственных препаратов».
23. Guideline on the investigation of drug interactions. EMA; 2012. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf
24. Решение Совета ЕЭК от 03.11.2016 № 89 «Об утверждении Правил проведения исследований биологических лекарственных средств Евразийского экономического союза».
25. Там же.
26. Персистенция ГТЛП — обнаружение векторных последовательностей или продукта трансгена в течение длительного времени после введения препарата.
27. Мобилизация — способность вируса / вирусной последовательности выходить из латентного состояния и реактивироваться вследствие непреднамеренной репликации после комплементации.
28. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
29. S12 Nonclinical biodistribution considerations for gene therapy products guidance for industry. ICH; 2023. https://database.ich.org/sites/default/files/ICH_S12_Step4_Guideline_2023_0314.pdf
30. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
31. Решение Совета ЕЭК от 03.11.2016 № 89 «Об утверждении Правил проведения исследований биологических лекарственных средств Евразийского экономического союза».
32. Решение Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
33. Guideline on similar biological medicinal products. EMA/CHMP/437/04. London; 2014.
Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance — quality issues. EMA/CHMP/BWP/247713/2012. London; 2014.
Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005. Rev. London; 2012.
1. Бем АЭ, Касимова АР, Гомон ЮМ, Колбин АС. Концепция мишень-опосредованного лекарственного распределения высокомолекулярных и низкомолекулярных соединений. Лекарственный вестник. 2021;15(3):3–12. EDN: XJMBJD
2. Rajput YK, Sahu TK. Pharmacokinetic consideration in drug development: A review. Hist Med. 2022;8(2):441–60.
3. Morgan P. The use of preclinical pharmacokinetic and pharmacodynamic data to predict clinical doses: current and future perspectives. International Congress Series. 2001;1220:1– 12. https://doi.org/10.1016/S0531-5131(01)00282-5
4. Жердев ВП, Бойко СС, Шевченко РВ, Гудашева ТА. Роль фармакокинетических и биофармацевтических исследований при создании новых дипептидных лекарственных средств (экспериментальное исследование). Фармакокинетика и фармакодинамика. 2017;(1):3–10. EDN: YPJQTV
5. Колыванов ГБ, Бочков ПО, Литвин АА и др. Абсолютная биодоступность соединения, обладающего кардиопротективной активностью (АЛМ-802), у крыс. Фармакокинетика и фармакодинамика. 2021;(2):31–5. https://doi.org/10.37489/2587-7836-2021-2-31-35
6. Абаимов ДА, Хуторова АВ, Сариев АК и др. Изучение базовых фармакокинетических свойств нового производного гистидин-содержащего дипептида карнозина — пирролилкарнозина. Фармакокинетика и фармакодинамика. 2023;(2):29–36. https://doi.org/10.37489/2587-7836-2023-2-29-36
7. Генатуллина ГН, Ясенявская АЛ, Цибизова АА, Самотруева МА. Нанокапсулированные системы: перспективные биомедицинские инициативы в фармакологии. Антибиотики и химиотерапия. 2024;69(3–4):62–72. https://doi.org/10.37489/0235-2990-2024-69-3-4-62-72
8. Yadav КS, Soni G, Choudhary D, et al. Microemulsions for enhancing drug delivery of hydrophilic drugs: Exploring various routes of administration. Med Drug Discov. 2023;20:100162. https://doi.org/10.1016/j.medidd.2023.100162
9. Haripriyaa M, Suthindhiran K. Pharmacokinetics of nanoparticles: Current knowledge, future directions and its implications in drug delivery. Futur J Pharm Sci. 2023;9:113. https://doi.org/10.1186/s43094-023-00569-y
10. Lai Y, Chu X, Di L, et al. Recent advances in the translation of drug metabolism and pharmacokinetics science for drug discovery and development. Acta Pharm Sin B. 2022;12(6): 2751–77. https://doi.org/10.1016/j.apsb.2022.03.009
11. Хушпульян ДМ, Гайсина ИН, Никулин СВ и др. Высокопроизводительный скрининг при поиске лекарств: проблемы и решения. Вecтник Московского университета. Серия 2: Химия. 2024;65(2):96–112. EDN: ARAUPF
12. Sandhya K, Ramesh Y, Penabaka V, Chandra YP. The role of pharmacokinetics in drug development. GSC Biol Pharm Sci. 2025;30(3):322–8. https://doi.org/10.30574/gscbps.2025.30.3.0098
13. Reichel A, Lienau P. Pharmacokinetics in drug discovery: An exposure-centred approach to optimising and predicting drug efficacy and safety. In: Nielsch U, Fuhrmann U, Jaroch S, eds. Handbook of Experimental Pharmacology. Springer; 2016. https://doi.org/10.1007/164_2015_26
14. Ruiz-Garcia A, Bermejo M, Moss A, Casabo VG. Pharmacokinetics in drug discovery. J Pharm Sci. 2008;97(2):654–90. https://doi.org/10.1002/jps.21009
15. White RE. High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery. Annu Rev Pharmacol Toxicol. 2000;40:133–57. https://doi.org/10.1146/annurev.pharmtox.40.1.133
16. Li Y, Meng Q, Yang M, et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm Sin B. 2019;9(6): 1113–44. https://doi.org/10.1016/j.apsb.2019.10.001
17. Rizk ML, Zou L, Savic RM, Dooley KE. Importance of drug pharmacokinetics at the site of action. Clin Transl Sci. 2017;10(3):133–42. https://doi.org/10.1111/cts.12448
18. Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. https://doi.org/10.1038/srep42717
19. Tuntland T, Ethell B, Kosaka T, et al. Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front Pharmacol. 2014;5:174. https://doi.org/10.3389/fphar.2014.00174
20. Zhang X, Zhou B, Gong Y, Liu Y. Investigation into the pharmacodynamics and pharmacokinetics of recombinant human interferon alfa-2b vaginal suppository following process optimization in Chinese rhesus macaque. Sci Rep. 2025; 15(1):15932. https://doi.org/10.1038/s41598-025-98813-3
21. Демина НБ, Бардаков АИ, Краснюк ИИ. Становление и развитие биофармацевтической доктрины создания эффективных лекарственных средств. Фармация. 2022;71(7):5–10. https://doi.org/10.29296/25419218-2022-07-01
22. Самойлов ВМ, Савицкий МВ, Ромашкина АГ и др. Исследование фармакокинетики твердодисперсной и кристаллической форм антивирулентного средства фтортиазинон у крыс. Медико-фармацевтический журнал «Пульс». 2023;25(7):57–62. https://doi.org//10.26787/nydha-2686-6838-2023-25-7-57-62
23. More SK, Pawar AP. Preparation, optimization and preliminary pharmacokinetic study of curcumin encapsulated turmeric oil microemulsion in zebra fish. Eur J Pharm Sci. 2020;155(1):105539. https://doi.org/10.1016/j.ejps.2020.105539
24. Ali SK, Al-Akkam EJ. Comparison of pharmacokinetic characteristics of bilosomal dispersion versus pure solution of oral ropinirole hydrochloride in rats. J Fac Med Baghdad. 2024;66(2): 201–8. https://doi.org/10.32007/jfacmedbagdad.6622210
25. Кравцова ОЮ, Дворянинов ДА, Колыванов ГБ и др. Экспериментальная фармакокинетика нового противопаркинсонического средства АДК-1113. Фармакокинетика и фармакодинамика. 2024;(1):27–31. https://doi.org/10.37489/2587-7836-2024-1-27-31
26. Бойко СС, Колясникова КН, Жердев ВП. Сравнительное изучение фармакокинетики и энзиматической устойчивости нейропротекторного средства ГЗК-111 и ноопепта у крыс. Фармакокинетика и фармакодинамика. 2022;(3):20–5. https://doi.org/10.37489/2587-7836-2022-3-20-25
27. Lu W, Zeng R, Pan M, et al. Pharmacokinetics, bioavailability, and tissue distribution of MRTX1133 in rats using UHPLC-MS/MS. Front Pharmacol. 2024;15:1509319. https://doi.org/10.3389/fphar.2024.1509319
28. Wilson SE, Carpenter JW, Gardhouse S, Kukanich B. Pharmacokinetics of mavacoxib in New Zealand White rabbits (Oryctolagus cuniculus). Am J Vet Res. 2023;84(5):ajvr.22.11.0196. https://doi.org/10.2460/ajvr.22.11.0196
29. Kumar PV, Maki MAA, Wei YS, et al. Rabbit as an animal model for pharmacokinetics studies of enteric capsule contains recombinant human keratinocyte growth factor loaded chitosan nanoparticles. Curr Clin Pharmacol. 2019;14(2):132–40. https://doi.org/10.2174/1574884714666181120103907
30. Шевченко РВ, Литвин АА, Колыванов ГБ и др. Фармакокинетика инъекционной лекарственной формы ГК-2 у кроликов. Фармакокинетика и фармакодинамика. 2020;(2):17–21. https://doi.org/10.37489/2587-7836-2020-2-17-21
31. Weir SJ, Wood R, Schorno K, et al. Preclinical pharmacokinetics of fosciclopirox, a novel treatment of urothelial cancers, in rats and dogs. J Pharmacol Exp Ther. 2019;370(2):148–59. https://doi.org/10.1124/jpet.119.257972
32. Xie H, Chung J-K, Mascelli MA, McCauley TG. Pharmacokinetics and bioavailability of a therapeutic enzyme (idursulfase) in cynomolgus monkeys after intrathecal and intravenous administration. PLoS One. 2015;10(4):e0122453. https://doi.org/10.1371/journal.pone.0122453
33. Енгалычева ГН, Сюбаев РД. Выбор релевантных видов животных для проведения доклинических исследований безопасности лекарственных средств: обзор. Безопасность и риск фармакотерапии. 2025;13(1):31–43. https://doi.org/10.30895/2312-7821-2025-460
34. Мирошников МВ, Султанова КТ, Макарова МН, Макаров ВГ. Сравнительный обзор активности ферментов системы цитохрома P450 человека и лабораторных животных. Прогностическая ценность доклинических моделей in vivo. Трансляционная медицина. 2022;9(5):44–77. https://doi.org/10.18705/2311-4495-2022-9-5-44-77
35. Heller AA, Lockwood SY, Janes TM, Spence DM. Technologies for measuring pharmacokinetic profiles. Annu Rev Anal Chem (Palo Alto Calif). 2018;11(1):79–100. https://doi.org/10.1146/annurev-anchem-061417-125611
36. Жердев ВП, Бойко СС, Шевченко РВ и др. Роль исследований межвидовых особенностей фармакокинетики в создании новых пептидных лекарственных средств. Фармакокинетика и фармакодинамика. 2018;(1):3–23. https://doi.org/10.24411/2587-7836-2018-10001
37. Zheng M-C, Tang W-T, Yu L-L, et al. Preclinical pharmacokinetics and bioavailability of oxypeucedanin in rats after single intravenous and oral administration. Molecules. 2022;27(11):3570. https://doi.org/10.3390/molecules27113570
38. Косман ВМ, Карлина МВ. Ретроспективная оценка вариабельности фармакокинетических параметров в зависимости от биологического вида и числа особей в экспериментальной группе. Лабораторные животные для научных исследований. 2023;6(1):60–9. https://doi.org/10.57034/2618723X-2023-01-06
39. Czerniak R. Gender-based differences in pharmacokinetics in laboratory animal models. Int J Toxicol. 2001;20(3):161–3. https://doi.org/10.1080/109158101317097746
40. Kushida H, Matsumoto T, Ikarashi Y, et al. Gender differences in plasma pharmacokinetics and hepatic metabolism of geissoschizine methyl ether from Uncaria hook in rats. J Ethnopharmacol. 2021;264:113354. https://doi.org/10.1016/j.jep.2020.113354
41. Arora P, Gudelsky G, Desai PB. Gender-based differences in brain and plasma pharmacokinetics of letrozole in Sprague-Dawley rats: Application of physiologically-based pharmacokinetic modeling to gain quantitative insights. PLoS One. 2021;16(4):e0248579. https://doi.org/10.1371/journal.pone.0248579
42. Смольякова ВИ, Чернышева ГА, Яновская ЕА и др. Оценка линейности фармакокинетики фенольного антиоксиданта 4-метил-2,6-диизоборнилфенола при внутрижелудочном введении. Экспериментальная и клиническая фармакология. 2014;77(2):31–4. EDN: SVVPPT
43. Кравцова ОЮ, Колыванов ГБ, Литвин АА и др. Оценка линейности фармакокинетики нейропротектора ГЗК-111. Экспериментальная и клиническая фармакология. 2023;86(3):8–11. https://doi.org/10.30906/0869-2092-2023-86-01-8-11
44. Anizan S, Concheiro M, Lehner KR, et al. Linear pharmacokinetics of 3,4-methylenedioxypyrovalerone (MDPV) and its metabolites in the rat: relationship to pharmacodynamic effects. Addict Biol. 2014;21(2):339–47. https://doi.org/10.1111/adb.12201
45. Shang H, Dai X, Li M, et al. Absolute bioavailability, dose proportionality, and tissue distribution of rotundic acid in rats based on validated LC-QqQ-MS/MS method. J Pharm Anal. 2022;12(2):278–86. https://doi.org/10.1016/j.jpha.2021.03.008
46. Колыванов ГБ, Литвин АА, Кравцова ОЮ и др. Фармакокинетика потенциального противоэпилептического средства ГИЖ-298 у крыс после различных режимов дозирования. Фармакокинетика и фармакодинамика. 2024;(2):57–61. https://doi.org/10.37489/2587-7836-2024-2-57-61
47. Cook CS, Zhang L, Ames GB, et al. Single- and repeated-dose pharmacokinetics of eplerenone, a selective aldosterone receptor blocker, in rats. Xenobiotica. 2003;33(3):305–21. https://doi.org/10.1080/0049825021000049277
48. Bochner F, Hooper WD, Tyrer JH, Eadie MJ. Factors involved in an outbreak of phenytoin intoxication. J Neurol Sci. 1972;16(4):481–7. https://doi.org/10.1016/0022-510x(72)90053-6
49. Silva DA, Löbenberg R, Davies N. Are excipients inert? Phenytoin pharmaceutical investigations with new incompatibility insights. J Pharm Pharm Sci. 2018;21(1s):29745. https://doi.org/10.18433/jpps29745
50. Thakur SK, Pal R, Pandey P, et al. Approaches of drug-excipients interaction in pharmaceutical drug product formulation. World J Pharm Res. 2023;12(2):347–66.
51. Gawari ShD, Kambale HV, Satpute VM, et al. A review: drug-excipient interactions study. Int J Novel Res Devel. 2023:8(2):b641–51.
52. Panakanti R, Narang AS. Impact of excipient interactions on drug bioavailability from solid dosage forms. Pharm Res. 2012:29(10):2639–59. https://doi.org/10.1007/s11095-012-0767-8
53. Ковальская ГН, Жукова ДЯ, Михалевич ЕН. Взаимодействие лекарственных средств для инъекционного и инфузионного применения. Сибирское медицинское обозрение. 2018;(6):12–21. https://doi.org/10.20333/2500136-2018-6-12-21
54. Логунова ИВ, Богомолова НС, Чистяков ВВ. Экспериментальное исследование биодоступности комбинированного препарата Диоксазид. Фармакокинетика и фармакодинамика. 2012;(1):29–32. EDN: RWVVCV
55. Yu DA, You M, Ji WW, et al. Preclinical pharmacokinetics of a recombinant humanized rabbit anti-VEGF monoclonal antibody in rabbits and monkeys. Toxicol Lett. 2018;292:73–7. https://doi.org/10.1016/j.toxlet.2018.04.031
56. Шерстобоев ЕЮ, Олейник ЛА, Жданов ВВ и др. Фармакокинетические параметры при пероральном введении пегилированного ИФН-l1. Бюллетень экспериментальной биологии и медицины. 2022;173(2):188–92. https://doi.org/10.47056/0365-9615-2022-173-2-188-192
57. Cai Y, Zhang Z, Fan K, et al. Pharmacokinetics, tissue distribution, excretion, and antiviral activity of pegylated recombinant human consensus interferon-α variant in monkeys, rats and guinea pigs. Regulatory Peptides. 2012;173(1–3): 74–81. https://doi.org/10.1016/j.regpep.2011.09.008
58. Смирнов ВВ, Петухова ОА, Филатов АВ и др. Исследование фармакокинетики биотехнологических препаратов на примере моноклональных антител. БИОпрепараты. Профилактика, диагностика, лечение. 2023;23(2):173–80. https://doi.org/10.30895/2221-996X-2023-23-2-173-180
59. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576–88. https://doi.org/10.1002/psp4.12224
60. Деркаев АА, Рябова ЕИ, Есмагамбетов ИБ и др. Кандидатный препарат на основе модифицированных однодоменных антител для терапии ботулизма, вызванного ботулиническим токсином типа А. БИОпрепараты. Профилактика, диагностика, лечение. 2025;25(1):58–70. https://doi.org/10.30895/2221-996X-2025-591
61. Астапова ОВ, Берчатова АА. Генотерапевтические препараты: аспекты доклинического изучения безопасности. Безопасность и риск фармакотерапии. 2023;11(1):73–96. https://doi.org/10.30895/2312-7821-2023-11-1-329
62. Копеин ДС, Порошин ГН, Хамитов РА. Реализация концепции Quality by Design для генотерапевтического лекарственного препарата на основе аденоассоциированного вирусного вектора. БИОпрепараты. Профилактика, диагностика, лечение. 2025;25(2):141–55. https://doi.org/10.30895/2221-996X-2025-580
63. Аляутдин РН, Романов БК, Переверзев АП и др. Алипоген типарвовек: долгая дорога к оценке отношения пользы и риска генетических препаратов. Ведомости Научного центра экспертизы средств медицинского применения. 2015;(1):31–4. EDN: UBDTJH
64. Рачинская ОА, Мельникова ЕВ, Меркулов ВА. Особенности производства и контроля качества соматотерапевтических лекарственных препаратов на основе мезенхимальных стволовых клеток. Антибиотики и химиотерапия. 2025;70(1–2):58–75. https://doi.org/10.37489/0235-2990-2025-70-1-2-58-75
65. Галицына ЕВ, Куликова ЕА, Павельев ЮА и др. Лекарственные препараты клеточной терапии: современное состояние исследований. БИОпрепараты. Профилактика, диагностика, лечение. 2024;24(4):428–42. https://doi.org/10.30895/2221-996X-2024-557
66. Wang X, Guo J, Deng X, et al. Evaluation of pharmacokinetics and toxicology of biosimilar APZ001 antibody in Macaca cynomolgus. Trop J Pharm Res. 2018;17(9):1885–91. https://doi.org/10.4314/tjpr.v17i9.30
67. Подолякина АИ, Дмитриева АА, Постникова ВА и др. Оценка фармакокинетики биоаналога пертузумаба в сравнении с оригинальным препаратом у яванских макак. Российский онкологический журнал. 2024;29(3):160–70. https://doi.org/10.17816/onco636526
68. Олефир ЮВ, Медуницын НВ, Авдеева ЖИ и др. Современные биологические/биотехнологические лекарственные препараты. Актуальные вопросы разработки и перспективы использования. БИОпрепараты. Профилактика, диагностика, лечение. 2016;16(2):67–77. EDN: WAIVUX
69. McLachlan AJ, Adiwidjaja J. Pharmacokinetics of biologics. In: Ramzan I, ed. Biologics, biosimilars, and biobetters: An introduction for pharmacists, physicians and other health practitioners. John Wiley & Sons; 2020. https://doi.org/10.1002/9781119564690.ch8
70. Максимкина ЕА, Кудрин A, Аладышева ЖИ и др. Государственное регулирование подобных биологических лекарственных препаратов для медицинского применения в Европейском Союзе. Ремедиум. 2013;(7–8):60–1. EDN: QZIHKN
71. Луговик ИА, Бабина АВ, Арутюнян СС и др. Первый дженерик тирзепатида GP30931: физико-химическое и биологическое сходство с референтным лекарственным средством. Разработка и регистрация лекарственных средств. 2025;14(2):54–74. https://doi.org/10.33380/2305-2066-2025-14-2-2084
72. Бредер ВВ. Биоаналоги в онкологии: новые горизонты, старые проблемы. Онкогематология. 2007;2(4):78–83. EDN: MSMSBN
Карлина Марина Валерьевна, канд. биол. наук, руководитель группы фармакокинетики, Группа научно-исследовательских институтов
Заводская ул., д. 3, к. 245, г.п. Кузьмоловский, Всеволожский р-н, Ленинградская обл., 188663
Косман Вера Михайловна, канд. фарм. наук
Заводская ул., д. 3, к. 245, г.п. Кузьмоловский, Всеволожский р-н, Ленинградская обл., 188663
Макарова Марина Николаевна, д-р мед. наук
Заводская ул., д. 3, к. 245, г.п. Кузьмоловский, Всеволожский р-н, Ленинградская обл., 188663
Макаров Валерий Геннадьевич, д-р мед. наук
Заводская ул., д. 3, к. 245, г.п. Кузьмоловский, Всеволожский р-н, Ленинградская обл., 188663
Карлина М.В., Косман В.М., Макарова М.Н., Макаров В.Г. Подходы к дизайну исследования фармакокинетики лекарственных средств в доклинических исследованиях (обзор). Регуляторные исследования и экспертиза лекарственных средств. 2026;16(2):163-178. https://doi.org/10.30895/1991-2919-2026-16-2-163-178
Karlina M.V., Kosman V.M., Makarova M.N., Makarov V.G. Approaches to Drug Pharmacokinetics Study Design in Preclinical Trials (Review). Regulatory Research and Medicine Evaluation. 2026;16(2):163-178. (In Russ.) https://doi.org/10.30895/1991-2919-2026-16-2-163-178

Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных




























