


ГЛАВНАЯ ТЕМА: ТРАНСФЕР МЕДИЦИНСКИХ ТЕХНОЛОГИЙ: ИНСТРУМЕНТЫ И МЕХАНИЗМЫ ДОСТИЖЕНИЯ ТЕХНОЛОГИЧЕСКОГО СУВЕРЕНИТЕТА В ЗДРАВООХРАНЕНИИ

Ключевая задача медицинской науки — развитие передовых технологий и внедрение инновационных продуктов, обеспечивающих сохранение и улучшение здоровья населения. Решение этой задачи требует всесторонней поддержки разработчиков на всех этапах жизненного цикла инноваций. Взаимодействие научного сообщества, государства и бизнеса в сфере практического здравоохранения позволит разработкам в области медицинской науки перейти от лабораторного стола к конкретному пациенту. О наиболее эффективных мерах поддержки и сопровождения перспективных проектов в медицине рассказала заместитель министра здравоохранения Российской Федерации Татьяна Владимировна Семенова.

ВВЕДЕНИЕ. Укрепление практического здравоохранения и повышение доступности фармакотерапии напрямую зависят от разработки и вывода в обращение инновационных отечественных препаратов. Среди инструментов вывода препаратов на рынок одним из важнейших является трансфер технологий.
ЦЕЛЬ. Оценка роли Центра трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России в исполнении мероприятий федеральных проектов в области медицинской науки и в достижении целей фармацевтического суверенитета.
ОБСУЖДЕНИЕ. В статье рассмотрены основные тенденции развития фармацевтической отрасли на национальном (в России) и международном уровне. Отмечены знаковые решения, принимаемые на федеральном уровне в целях достижения фармацевтического суверенитета и повышения доступности фармакотерапии для населения. Рассмотрено Положение о деятельности Центра трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России, очерчен круг его полномочий, проанализированы возможности по сопровождению всех этапов проектов разработки лекарственных препаратов, в том числе по вопросам эффективности деятельности по разработке лекарственных препаратов, вопросам патентного права, а также коммерциализации продукта. Оценена роль Центра трансфера медицинских технологий в развитии фармацевтической отрасли в Российской Федерации.
ВЫВОДЫ. Деятельность Центра трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России по сопровождению проектов осуществляется в целях исполнения отдельных мероприятий федеральных проектов в области медицинской науки для человека и может являться одним из инструментов, способствующих эффективному инновационному развитию отечественной фармацевтической отрасли.

ВВЕДЕНИЕ. Коммерческий успех инновационных разработок в медицине и фармации напрямую зависит от стратегического планирования патентной охраны, основными элементами которого являются режим патентной правовой охраны, объект изобретения, последовательность получения охранных документов.
ЦЕЛЬ. Анализ особенностей стратегического планирования патентной охраны фармацевтической разработки.
МАТЕРИАЛЫ И МЕТОДЫ. В исследовании применялись аналитический и сравнительный методы исследования патентной информации. Для подготовки статистических данных использовались патентные базы данных: поисковая платформа Роспатента и Questel.
РЕЗУЛЬТАТЫ. В Российской Федерации количество патентов на изобретения, относящихся к способам лечения и диагностики, превалирует над патентами, относящимися к фармацевтическому продукту. Наиболее рациональным способом охраны права интеллектуальной собственности в фармацевтической отрасли является поэтапное получение разного вида охранных документов в рамках одного проекта на различные объекты патентного права: изобретения, полезные модели, промышленные образцы. При построении стратегии патентной охраны фармацевтической разработки следует учитывать: выбор режима патентной охраны (охрана разработки одновременно с использованием патентов на изобретения, полезные модели и промышленные образцы); корректное определение объекта патентования (получение патента на «продукт» (вещество)); поэтапное получение разного вида охранных документов и наращивание их.
ВЫВОДЫ. Представленная стратегия позволяет в наибольшей степени охватить все возможные варианты охраны прав на разработку и предупредить нарушения, а также удлинить срок правовой охраны разработки.
ВВЕДЕНИЕ. Исследование отношений в области государственной поддержки инновационного развития отрасли здравоохранения, в частности института государственно-частного партнерства, приобретает особую актуальность на фоне формирования технологического суверенитета при сложившейся геополитической ситуации. В условиях санкций и экономического кризиса, возможного дефицита национальных инновационных медицинских продуктов, устойчиво конкурирующих с экспортными, российское государство сталкивается с необходимостью финансирования разработок и производства инновационных медицинских продуктов, в том числе с привлечением частных партнеров (инвесторов). При формировании технологического суверенитета необходимо привлечение механизмов государственно-частного партнерства, что позволит увеличить приток частного капитала и компетенций и может существенно повлиять на сроки разработки, реализации и внедрения инновационных медицинских продуктов, улучшить качество медицинской помощи и производство лекарственных средств и медицинских изделий.
ЦЕЛЬ. Анализ существующих механизмов государственно-частного партнерства для развития отечественной сферы здравоохранения и научно-технической отрасли в рамках взаимодействия государство — бизнес и выявление оптимальных моделей при разработке, реализации, внедрении и производстве инновационных медицинских продуктов.
МАТЕРИАЛЫ И МЕТОДЫ. В исследовании использовался информационно-аналитический подход для определения наиболее перспективных механизмов инновационного развития здравоохранения с акцентом на модели государственно-частного партнерства. Объектом исследования являлось федеральное законодательство Российской Федерации.
РЕЗУЛЬТАТЫ. В статье рассмотрены часто встречающиеся формы возможного участия и поддержки государством инновационного развития здравоохранения с целью роста эффективности и масштабирования реализации и внедрения инновационных медицинских продуктов. Рассмотрено применение финансирования проектов технологического суверенитета, расширение сферы действия преференциальных режимов и концепция применения модели государственно-частного партнерства. В статье представлены механизмы реализации государственно-частного партнерства через различные формы, включая специальные инвестиционные контракты, концессионные соглашения, соглашения о государственно-частном партнерстве. Эти механизмы позволяют передавать объекты инфраструктуры в управление частным партнерам, что может включать производственные мощности, исследовательские центры и другие объекты, необходимые для инновационного развития. Особое внимание уделено рассмотрению условий и особенностей концессионных соглашений в сфере здравоохранения, таких как распределение ответственности и обязанностей сторон, применение риск-ориентированного подхода и выявление в сложившейся деловой практике необходимых для сторон условий.
ВЫВОДЫ. При реализации государственно-частных партнерств в сфере инновационного развития отрасли здравоохранения необходимо учитывать и преодолевать сдерживающие факторы, в том числе технологические и административные барьеры, которые могут препятствовать эффективному развитию. Наиболее предпочтительной формой реализации государственно-частного партнерства является, по мнению авторов, модель концессионного соглашения.
РЕГУЛИРОВАНИЕ ОБРАЩЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ

ВВЕДЕНИЕ. Степень обработки (манипулирования) клеток, входящих в состав клеточных препаратов, и выполняемые ими функции после введения (гомологичное/ негомологичное применение) определяют их отнесение к классу трансплантатов или высокотехнологичных лекарственных препаратов и, соответственно, особенности регулирования их обращения. В законодательстве Евразийского экономического союза (ЕАЭС) и Российской Федерации в настоящее время недостаточно полно разъяснены термины «минимальные манипуляции» и «гомологичное/негомологичное применение», что может служить причиной применения у человека клеточных препаратов с недоказанной безопасностью и эффективностью.
ЦЕЛЬ. Сравнение международных и российских подходов к интерпретации терминов «минимальные манипуляции», «гомологичное/негомологичное применение» на примере препаратов стромально-васкулярной фракции (СВФ) с целью классификации препарата и определения пути его регулирования.
ОБСУЖДЕНИЕ. Рассмотрены и обобщены регуляторные подходы Российской Федерации, ЕАЭС, США и ЕС в зависимости от классификации клеточных препаратов по степени манипулирования входящих в их состав клеток и выполняемых ими функций после применения. Проведены анализ и сравнение нормативно-правовых актов и подходов рассмотренных стран на примере препаратов на основе СВФ. Показаны общие аспекты толкования терминов «минимальные манипуляции» и «гомологичное/негомологичное применение», продемонстрировано различие в регуляторных подходах разных стран, заключающееся в отнесении ферментативной обработки и селективного отбора клеток к существенным или минимальным манипуляциям.
ВЫВОДЫ. Механизм регулирования обращения клеточных препаратов зависит от степени обработки клеток в их составе (минимальное или существенное манипулирование) и от предполагаемого применения (гомологичного или негомологичного). Общим принципом отнесения манипуляций к минимальным, принятым регуляторными организациями США, ЕС, ЕАЭС, Российской Федерации, является сохранение биологических характеристик и физиологической функции продуктов после их проведения; признаком гомологичного применения клеток или тканей является их использование для выполнения присущих им функций в организме. В России перечень манипуляций, отнесенных к минимальным, определен в нормативных актах как для высокотехнологичных лекарственных препаратов, так и для объектов трансплантации. Препараты на основе минимально манипулированных клеток стромально-васкулярной фракции, согласно международным нормам, при негомологичном применении классифицируются как высокотехнологичные лекарственные препараты. Термины «минимальные манипуляции» и «гомологичное/негомологичное применение» недостаточно полно и четко определены в законодательстве ЕАЭС и Российской Федерации, поэтому актуальной является разработка рекомендаций по их разъяснению на конкретных примерах.
КОНТРОЛЬ КАЧЕСТВА
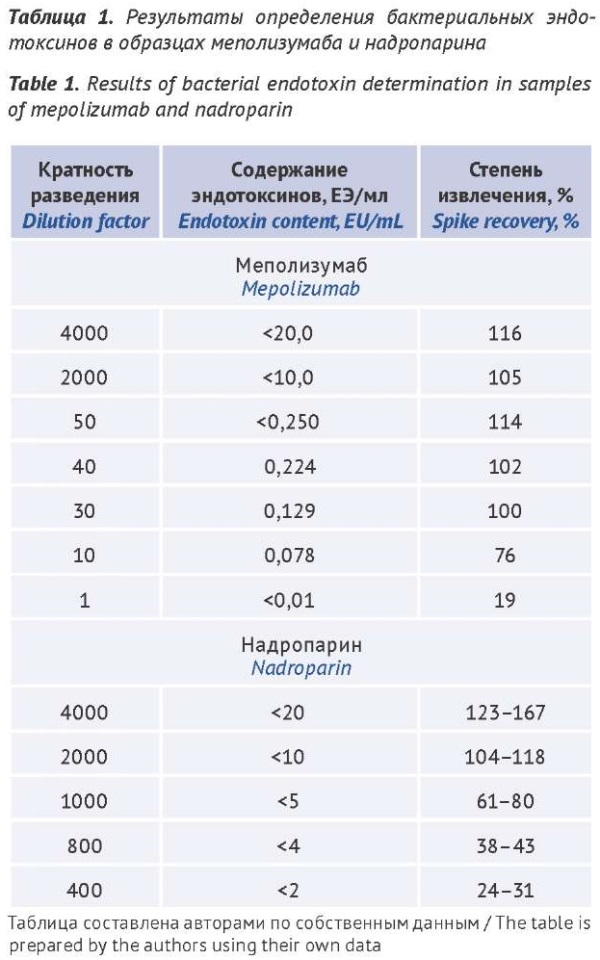
ВВЕДЕНИЕ. Кинетические методы определения бактериальных эндотоксинов (БЭ) в лекарственных препаратах для парентерального применения с помощью лизата амебоцитов из гемолимфы мечехвостов позволяют количественно определять содержание эндотоксинов в широком диапазоне концентраций. Для разработки конкретной методики на основе этих методов для контроля качества и стандартизации лекарственных препаратов необходимо уточнить условия ее использования, в частности установить наименьшее разведение, в котором отсутствуют мешающие факторы.
ЦЕЛЬ. Разработка методики количественного определения бактериальных эндотоксинов хромогенным кинетическим методом.
МАТЕРИАЛЫ И МЕТОДЫ. Испытывали растворы образцов лекарственных препаратов с действующими веществами различных фармакологических групп: моноклональные антитела — «меполизумаб» и антикоагулянт прямого действия — «надропарин», с нормами предельного содержания БЭ «не более 20 ЕЭ/мл» и «не более 95 ЕЭ/мл» соответственно, с разной кратностью разведения, но не превышающей значения максимально допустимого разведения. Исследование выполняли с использованием фотоколориметра «BioТek EL×800» и программного обеспечения «Endoscan-V».
РЕЗУЛЬТАТЫ. В исследовании препарата с действующим веществом «меполизумаб» установлено, что в диапазоне разведений от 1:10 до 1:40 хромогенным кинетическим методом возможно обнаружить БЭ в количествах 0,0078–0,224 ЕЭ/мл. Начиная с разведения 1:50 метод позволяет осуществить только качественную оценку наличия БЭ. Содержание БЭ в лекарственном препарате с действующим веществом «надропарин» следует определять начиная с разведения 1:1000, в котором влияние мешающих факторов отсутствует.
ВЫВОДЫ. Разработаны методики определения БЭ хромогенным кинетическим методом (метод D) для препаратов меполизумаба и надропарина. Рекомендовано при разработке методик количественного определения БЭ в индивидуальных лекарственных препаратах определять наименьшее разведение образца, в котором отсутствуют мешающие факторы, и вносить эту информацию в документацию методики. Данный подход повышает точность выполнения анализа, обеспечивает экономию материальных ресурсов и времени при проведении рутинных испытаний.
ВВЕДЕНИЕ. Использование биологического сырья при получении лекарственных средств (ЛС) является причиной возможного загрязнения продукта депрессорными примесями, такими как гистамин, ацетилхолин, брадикинин, серотонин, простагландины и т.п. На территории Российской Федерации контроль качества ЛС по содержанию таких примесей проводят в соответствии с фармакопейным биологическим методом «Испытание на депрессорные вещества», которое выполняют на здоровых наркотизированных кошках. В связи с общемировой тенденцией исключения испытаний на животных рассмотрены подходы ведущих стран к определению гистаминоподобных веществ.
ЦЕЛЬ. Анализ российских и международных подходов к контролю содержания депрессорных веществ в биологических ЛС. Рассмотрение возможности применения инструментальных физико-химических методов определения гистаминоподобных примесей в качестве альтернативных биологическим методам с целью сокращения количества испытаний на животных.
ОБСУЖДЕНИЕ. Причиной сокращения применения животных в качестве тест-системы в испытаниях послужила Директива Европейского парламента и Совета Европейского союза 2010/63/ЕС от 22 сентября 2010 г. о защите животных, используемых для научных целей. В 2023 г. эксперты Европейской фармакопейной комиссии приняли решение о начале работ по удалению из фармакопеи показателей in vivo для определения содержания гистаминоподобных примесей. В Индии и Китае испытание на животных применяют в качестве основного для определения безопасности ЛС. На территории Российской Федерации проведение испытания на депрессорные вещества регламентируется ОФС 1.2.4.0008.18 Государственной фармакопеи Российской Федерации (ГФ РФ). С 2007 г. в ГФ РФ XII изд. включена ОФС 42-0063-07 «Испытание на гистамин» с целью контроля содержания гистамина в ЛС, которая позволяет сократить количество используемых в экспериментах кошек. В настоящее время российскими исследователями ведется поиск однонаправленных физико-химических или иммунохимических методов определения депрессорных веществ. Одним из путей решения этой проблемы может быть индивидуальный подход к назначению испытания для каждого контролируемого ЛС, что позволит минимизировать количество проводимых испытаний in vivo при сохранении надлежащего качества ЛС.
ВЫВОДЫ. Несмотря на общемировую тенденцию к сокращению применения метода in vivo, контроль содержания депрессорных веществ необходим и востребован. В мировом сообществе отсутствует единое мнение о необходимости использования животных для оценки депрессорных веществ. Отсутствие упоминания испытания в фармакопее как отдельной статьи (монографии) не отменяет факта повсеместного контроля гистаминоподобных примесей. Решение об исключении методов in vivo из Европейской фармакопеи является стимулом для поиска новых аналитических методов и внедрения дополнительных надзорных мер при производстве ЛС, которые позволят минимизировать риск загрязнения готовой продукции примесями. Рассмотрена возможность применения высокоэффективной жидкостной хроматографии для определения содержания примеси гистамина в ограниченном количестве ЛС.

ВВЕДЕНИЕ. В связи с изданием в 2023 г. нового руководства Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения ICH Q14 «Разработка аналитических методик» и пересмотром руководства ICH Q2 по валидации аналитических методик используемый в настоящее время на территории стран ЕАЭС подход к разработке и валидации аналитических методик должен быть в значительной степени пересмотрен. Также подлежат пересмотру процедуры оценки значимости и внесения изменений в регистрационное досье, касающееся аналитических методик.
ЦЕЛЬ. Анализ существенных изменений международных подходов к разработке аналитических методик, оценка их преимуществ и недостатков при внедрении фармацевтическими предприятиями и регуляторными органами стран Евразийского экономического союза.
ОБСУЖДЕНИЕ. Рассмотрены основные положения и практические аспекты введенного руководством ICH Q14 расширенного подхода к разработке аналитических методик, включая концепции жизненного цикла аналитической методики (ЖЦАМ) и модифицированного подхода «качество через дизайн» (AQbD), разработку аналитического целевого профиля, управление рисками для качества испытаний, планирование экспериментов и стратегии контроля, валидацию аналитической методики и последующую верификацию, трансфер и управление изменениями аналитической методики. Также определены сопутствующие этой новой регуляторной парадигме обновления руководства ICH Q2 (R2).
ВЫВОДЫ. Указанные руководства восполняют дефицит рекомендаций в отношении разработки аналитических методик, а использование концепций ЖЦАМ и AQbD обеспечивает гибкость подходов как для фармацевтических компаний, так и для регуляторных органов. Эти подходы применимы как к новым разрабатываемым аналитическим методикам, так и к уже используемым на предприятиях. Для их эффективного внедрения в российскую фармацевтическую отрасль и регуляторную систему требуется широкая дискуссия специалистов и экспертов регуляторных органов, возможно, в рамках пилотного проекта, последующее обучение специалистов, занимающихся аналитической разработкой, а также внесение изменений в Правила регистрации и экспертизы лекарственных средств для медицинского применения.
ФАРМАКОПЕЯ

ВВЕДЕНИЕ. Регулярный анализ номенклатуры общих фармакопейных и фармакопейных статей является необходимым для обеспечения сбалансированного развития Государственной фармакопеи Российской Федерации (ГФ РФ) в соответствии с приоритетами отечественного здравоохранения.
ЦЕЛЬ. Определить перспективные направления развития Государственной фармакопеи Российской Федерации на основании анализа изменений российского законодательства и номенклатуры фармакопейных статей.
ОБСУЖДЕНИЕ. Проведен анализ текущего состояния ГФ РФ XV изд. в сравнении с ГФ РФ XIV изд. Рассмотрены ключевые особенности и структура ГФ РФ XV изд., изменения нормативных правовых актов, регулирующих ее издание. Анализ перечня фармакопейных статей (ФС) показал преобладание ФС на фармацевтические субстанции синтетического происхождения, наличие значительной доли статей на лекарственное растительное сырье и фармацевтические субстанции растительного происхождения. Отмечено, что в ГФ РФ XV изд. впервые введен раздел со статьями на вспомогательные вещества, которые необходимы для развития изготовления лекарственных препаратов в условиях аптек. Проведен анализ охвата фармакопейными требованиями субстанций, используемых для производства жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП).
ВЫВОДЫ. Обозначены приоритетные направления развития стандартизации качества: продолжение разработки ФС на фармацевтические субстанции, используемые для производства и изготовления лекарственных препаратов из перечня ЖНВЛП; расширение номенклатуры ФС на лекарственное растительное сырье; разработка ФС на вспомогательные вещества, применяемые для производства и изготовления лекарственных препаратов; завершение разработки требований к биологическим лекарственным препаратам и методам их анализа.
ЛЕКАРСТВЕННЫЕ СРЕДСТВА РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ
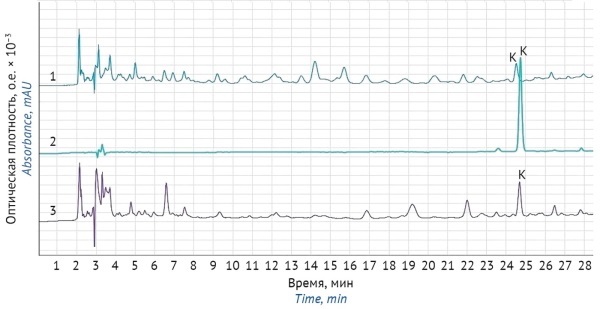
ВВЕДЕНИЕ. Надлежащий контроль содержания сердечных гликозидов в лекарственных препаратах на основе ландыша необходим для обеспечения безопасности их применения. В Российской Федерации фармакопейными методами количественного анализа сердечных гликозидов ландыша являются биологический и спектрофотометрический. Необходимо использовать более точные и селективные физико-химические методы анализа сердечных гликозидов, например метод высокоэффективной жидкостной хроматографии (ВЭЖХ).
ЦЕЛЬ. Разработка и валидация методики количественного определения сердечных гликозидов (конваллятоксина) в лекарственных растительных препаратах на основе ландыша методом ВЭЖХ.
МАТЕРИАЛЫ И МЕТОДЫ. В качестве объектов исследования были использованы: «Ландыша настойка», «Зеленина капли», «Валокормид», «Карниланд®», «Экстракт ландыша — стандартный образец». Количественное определение сердечных гликозидов проводили методами ВЭЖХ и спектрофотометрии в сравнении со стандартным образцом конваллятоксина. Для пробоподготовки использовали методику, описанную в ФС.3.4.0003.18. «Ландыша травы настойка». Хроматографическое разделение смеси сердечных гликозидов проводили на колонке Luna 5µm C18(2) в градиентном режиме элюирования смесью 0,1% раствора ортофосфорной кислоты и ацетонитрила. В процессе анализа использовали автосемплер с охлаждением проб до 5 °С.
РЕЗУЛЬТАТЫ. Результаты оценки специфичности, внутрилабораторной прецизионности, линейности (коэффициент корреляции 0,99985), правильности (98–102%), повторяемости (относительное стандартное отклонение содержания конваллятоксина — 1,61%) методики соответствуют критериям приемлемости. Методика пригодна для количественного определения конваллятоксина в лекарственных препаратах на основе ландыша, так как позволяет получать достоверные и воспроизводимые результаты.
ВЫВОДЫ. Разработана высокочувствительная и селективная ВЭЖХ-методика количественного определения конваллятоксина в лекарственных растительных препаратах на основе ландыша. Содержание конваллятоксина в субстанции-жидкости «Ландыша настойка» составило от 0,012 до 0,018 мг/мл, в лекарственных препаратах «Зеленина капли» — от 0,004 до 0,013 мг/мл, «Карниланд®» и «Валокормид» — от 0,005 до 0,007 мг/мл, в экстракте ландыша — стандартном образце — 0,029 мг/мл.
КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
ВВЕДЕНИЕ. В процессе экспертизы протоколов клинических исследований лекарственных препаратов местного действия (ЛПМД) выявляются затруднения, возникающие у разработчиков препаратов при выборе дизайна исследований, конечных точек, популяции, групп сравнения и размера выборки. Анализ типичных ошибок в протоколах клинических исследований позволит сократить количество замечаний при экспертизе и ускорит вывод новых препаратов на фармацевтический рынок.
ЦЕЛЬ. Проанализировать результаты экспертизы клинических исследований ЛПДМ, проведенных с учетом дополнений к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза (ЕАЭС), оценить основные преимущества введения дополнительных требований и сохраняющиеся трудности в разработке протоколов, представить рекомендации, позволяющие наиболее эффективно применять существующие нормативные правовые требования.
ОБСУЖДЕНИЕ. С момента вступления в силу Приложений № 11, 12 и 13 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках ЕАЭС в августе 2023 г. накоплен достаточный опыт проведения экспертиз протоколов клинических исследований ЛПМД. При анализе протоколов клинических исследований выявлены наиболее часто встречающиеся замечания и предложены рекомендации по их устранению. Отмечено, что затруднения при разработке протоколов исследований ЛПМД чаще всего вызывают выбор и обоснование первичных и вторичных конечных точек, определение популяции исследования, обоснование численности. Показано, что возможная причина данных затруднений, вероятно, связана с отсутствием подробного описания характеристик исследования в нормативных правовых актах ЕАЭС. При этом отмечено, что наименьшее количество трудностей вызывает разработка протоколов исследования кортикостероидных лекарственных препаратов для местного применения, описанная наиболее полно в законодательстве ЕАЭС и научной литературе.
ВЫВОДЫ. Проведенный анализ протоколов клинических исследований ЛПМД и соответствующих требований ЕАЭС и международных руководств позволил определить основные проблемы, связанные с выбором первичных и вторичных конечных точек исследования, характеристикой популяции и обоснованием ее численности. Представленные рекомендации помогут заявителям в планировании клинических исследований с целью ускорения вывода лекарственных препаратов на фармацевтический рынок.
ВВЕДЕНИЕ. Биоаналитические лаборатории в Российской Федерации должны одновременно соответствовать как требованиям надлежащей лабораторной практики (GLP) для проведения доклинических исследований, так и требованиям надлежащей клинической практики (GCP) для анализа образцов клинических исследований. Для работы подобных лабораторий необходимо создание отдельной ниши GxP практик — принципов надлежащей клинической лабораторной практики (GCLP). Однако данная область регулирования на территории Российской Федерации и стран Евразийского экономического союза (ЕАЭС) на данный момент не сформирована.
ЦЕЛЬ. Сравнительный анализ существующего положения в области регулирования деятельности биоаналитических лабораторий на территории Российской Федерации и стран ЕАЭС с международными принципами надлежащей клинической лабораторной практики для формирования общих требований к работе отечественных лабораторий.
ОБСУЖДЕНИЕ. Проанализированы текущее положение в области регулирования деятельности биоаналитических лабораторий, нормативные документы ЕАЭС по разработке и валидации аналитических методик, а также международные принципы GCLP, включающие кадровую политику лабораторий, порядок документирования процессов, разработку стандартных операционных процедур, валидацию методик, управление биологическими и стандартными образцами. Рассмотрены основные виды деятельности биоаналитических лабораторий и предложены инструменты управления ими, соответствующие международным принципам и требованиям ЕАЭС. Отмечено, что мировая практика применения принципов GCLP достаточно хорошо описана в научной литературе, и этот опыт может быть полезен для применения в работе отечественных лабораторий.
ВЫВОДЫ. Внедрение принципов GCLP в деятельность биоаналитических лабораторий в странах ЕАЭС приведет к повышению конкурентоспособности лаборатории на рынке биоаналитических услуг и снижению риска получения недостоверных данных.

ISSN 3034-3453 (Online)





























