



Содержание
Перейти к:
 А. В. Степанова,
А. Э. Тихонова,
А. С. Кошевенко,
Н. В. Попов,
К. Ю. Беланов,
А. А. Трапкова,
В. Ю. Емельянов,
С. В. Суров,
О. А. Мансуров,
К. А. Хрянин,
К. Ю. Казарян
А. В. Степанова,
А. Э. Тихонова,
А. С. Кошевенко,
Н. В. Попов,
К. Ю. Беланов,
А. А. Трапкова,
В. Ю. Емельянов,
С. В. Суров,
О. А. Мансуров,
К. А. Хрянин,
К. Ю. Казарян https://doi.org/10.30895/1991-2919-2025-15-4-391-403
Перейти к:
ВВЕДЕНИЕ. Потребность в новых решениях для диагностики и лечения широкого спектра заболеваний с использованием радиофармацевтических лекарственных препаратов (РФЛП), а также растущий рынок ядерной медицины стимулируют активную разработку и быстрый вывод в обращение инновационных РФЛП. Особенностью этого типа препаратов является наличие многокомпонентной структуры и специфической радиационной активности, что необходимо учитывать при планировании и контроле исследований по разработке препарата и дальнейшем трансфере технологий.
ЦЕЛЬ. Оптимизация процесса разработки РФЛП с помощью отраслевой шкалы уровней готовности технологии.
ОБСУЖДЕНИЕ. Рассмотрено применение отраслевой шкалы уровней готовности технологии РФЛП. Данная шкала учитывает ключевые особенности РФЛП: содержание в составе радиоактивного изотопа и биологической молекулы; необходимость контроля специфических показателей качества (радиохимической чистоты, стабильности и биологической активности), а также особенности обращения РФЛП на рынке. Критически важным является определение варианта применения и обращения разрабатываемого РФЛП, так как это определяет не только стратегию регистрации, но и требования к показателям качества получаемого препарата. На основании опыта АО «Росатом Наука» отмечены ключевые риски обращения (контаминация производственного оборудования, учет скорости радиоактивного распада при логистике и др.). Обсуждены особенности разработки воспроизведенных РФЛП (ускоренное прохождение первых четырех этапов разработки, необходимость подбора оригинальной молекулы-мишени), а также дизайн доклинических и клинических исследований для подтверждения биоэквивалентности воспроизведенного и референтного препаратов.
ВЫВОДЫ. Адаптированная шкала уровней готовности технологии РФЛП является инструментом, позволяющим комплексно контролировать ключевые этапы разработки, своевременно выявлять и минимизировать риски. Использование шкалы способствует ускорению вывода инновационных и воспроизведенных РФЛП на рынок за счет рутинного мониторинга процесса и поэтапного планирования полного цикла разработки, вплоть до стратегии обращения РФЛП на рынке.
Степанова А.В., Тихонова А.Э., Кошевенко А.С., Попов Н.В., Беланов К.Ю., Трапкова А.А., Емельянов В.Ю., Суров С.В., Мансуров О.А., Хрянин К.А., Казарян К.Ю. Планирование и управление процессами разработки радиофармацевтических препаратов с применением шкалы уровней готовности технологии. Регуляторные исследования и экспертиза лекарственных средств. 2025;15(4):391-403. https://doi.org/10.30895/1991-2919-2025-15-4-391-403
Stepanova A.V., Tikhonova A.E., Koshevenko A.S., Popov N.V., Belanov K.Yu., Trapkova A.A., Emelyanov V.Yu., Surov S.V., Mansurov O.A., Khryanin K.A., Kazaryan K.Yu. Planning and Management of Radiopharmaceutical Development Processes Using Technology Readiness Level Scale. Regulatory Research and Medicine Evaluation. 2025;15(4):391-403. (In Russ.) https://doi.org/10.30895/1991-2919-2025-15-4-391-403
Российский рынок радиофармацевтических препаратов (РФЛП) активно развивается в рамках государственной стратегии по укреплению технологической независимости страны и развитию ядерной медицины. По словам министра здравоохранения М.А. Мурашко, в настоящее время в России происходит взрывной рост разработок РФЛП и их применения1.
Программа «Новые технологии сбережения здоровья» предусматривает разработку и вывод на рынок к 2030 г. не менее 8 инновационных РФЛП2, а также достижение на российском рынке доли 95% РФЛП, производство которых локализовано в Российской Федерации3. С целью определения текущего объема производства и уровня обеспечения медицинских учреждений, анализа потребностей отрасли и возможностей отечественного рынка, оценки потенциала импортозамещения и перспектив экспорта РФЛП Минздрав России в рамках стратегического партнерства с Государственной корпорацией «Росатом» (ГК «Росатом») запустил проект «Инцидент-12», в ходе реализации которого создается комплексная карта производителей РФЛП4.
Важно отметить, что РФЛП принципиально отличаются от лекарственных средств, не содержащих радиоизотопы, по многим параметрам, включая условия и сроки хранения, особенности изготовления, производства, обращения, процесса разработки. Современные РФЛП обладают двойственной природой: они объединяют в своем составе химические/биологические субстанции и источники ионизирующего излучения [1][2]. Трансфер технологий изготовления/производства РФЛП имеет некоторые особенности, в том числе в части планирования разработки на всех этапах, включая проведение доклинических (ДКИ) [3][4] и клинических (КИ) исследований [5][6], и должен учитывать специальные требования регуляторных органов [7]. Вид обращения РФЛП на рынке (изготовление и (или)производство), а также форма реализации препарата является принципиально важным, в частности, для выбора регистрационной стратегии.
Производители РФЛП могут быть разделены на две группы: изготовители РФЛП в масштабе медицинской организации, включая ядерные аптеки, выпускающие лекарственные препараты с учетом индивидуальных потребностей пациентов; крупные производители медицинских изотопов и РФЛП. К группе изготовителей относятся прежде всего медицинские учреждения, подведомственные Минздраву России, которые одновременно являются и разработчиками РФЛП, например ФГБУ «Национальный медицинский исследовательский центр радиологии», ФГБУ «Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина», ФГБУ «Национальный медицинский исследовательский центр онкологии им. Н.Н. Петрова», ФГБУ «Российский научный центр радиологии и хирургических технологий им. академика А.М. Гранова», ФГБУ «Национальный медицинский исследовательский центр им. В.А. Алмазова». Кроме того, необходимо также отметить организации, входящие в ФМБА России: ФГБУ «Федеральный научно-клинический центр медицинской радиологии и онкологии» и ФГБУ «ГНЦ Федеральный медицинский биофизический центр им. А.И. Бурназяна».
Группа производителей представлена подразделениями, входящими в состав Государственной корпорации «Росатом» (например, АО «Научно-исследовательский физико-химический институт им. Л.Я. Карпова», ФГУП «ПО «Маяк», АО «Государственный научный центр Научно-исследовательский институт атомных реакторов», АО «Институт реакторных материалов», АО «Радиевый институт им. В.Г. Хлопина») и ФГУП «Федеральный центр по проектированию и развитию объектов ядерной медицины» ФМБА России. ГК «Росатом» входит в пятерку крупнейших мировых поставщиков сырьевой изотопной продукции и является ключевым поставщиком изотопной продукции медицинского назначения на российском рынке, обеспечивая РФЛП более 180 медицинских учреждений в Российской Федерации. Специалисты подразделения ГК «Росатом» АО «Росатом Наука» обладают уникальным опытом в сфере разработки РФЛП.
Разработка любого лекарственного средства — это сложный наукоемкий технологический процесс, сопряженный с высоким риском недостижения поставленного результата, что в том числе связано с отсутствием комплексного понимания жизненного цикла продукта у разработчиков на ранних стадиях исследования и внимания к критическим параметрам разрабатываемой молекулы. В целях снижения рисков для проектов по созданию лекарственных препаратов используются такие подходы, как, например, «качество через дизайн» (Quality-by-Design, QbD)5 [8]. QbD может быть эффективно применен при проведении исследований, однако в настоящее время нет четко описанных алгоритмов для разработки отдельных лекарственных форм и групп препаратов [9], также данная система не является прозрачной для заказчика, исследователя и регулирующего органа. При этом исследователи уже используют систему QbD, в частности для разработки препаратов на основе 99mTc [10].
Еще одним инструментом для планирования исследования и оценки прогресса при создании лекарственного препарата является шкала уровней готовности технологий (УГТ), которая позволяет оперативно выявлять вероятные риски невозможности достижения желаемых результатов разработки при ее продвижении от этапа формирования гипотезы до выхода на рынок [9][11]. Центром трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России (ЦТМТ) с учетом требований нормативных правовых актов и концепции QbD разработаны отраслевые УГТ [12], в том числе шкала, специфичная для РФЛП. В настоящее время данные отраслевые УГТ уже размещены в отраслевом сегменте Единой государственной информационной системы учета научно-исследовательских, опытно-конструкторских и технологических работ гражданского назначения (ЕГИСУ НИОКТР) домена «Наука и инновации» и активно используются разработчиками лекарственных препаратов, в частности в АО «Росатом Наука» в практике проведения исследований в области ядерной медицины в рамках комплексного проектного подхода при сопровождении исследований инновационных РФЛП.
Цель работы — оптимизация процесса разработки радиофармацевтических лекарственных препаратов с помощью отраслевой шкалы уровней готовности технологий.
Задачи исследования: детальный разбор отраслевой шкалы УГТ для РФЛП; выделение особенностей применения шкалы УГТ при планировании и мониторинге ключевых стадий разработки оригинальных и воспроизведенных РФЛП; анализ опыта разработчика РФЛП АО «Росатом Наука».
Шкала УГТ для РФЛП, подготовленная ЦТМТ, так же как и основная отраслевая шкала УГТ для лекарственных препаратов [12], представляет собой последовательное описание 9 уровней готовности с указанием обязательных, желательных и необязательных работ для каждого уровня (рис. 1). Оценка текущего значения УГТ определяется степенью реализации планируемых и проводимых работ от 1 (начальный уровень) до 9 (зрелый уровень). Уровень готовности технологии считается достигнутым при выполнении всех обязательных ключевых этапов работ и наличии документального подтверждения каждого результата согласно базовому рубрикатору научных и (или) научно-технических результатов.
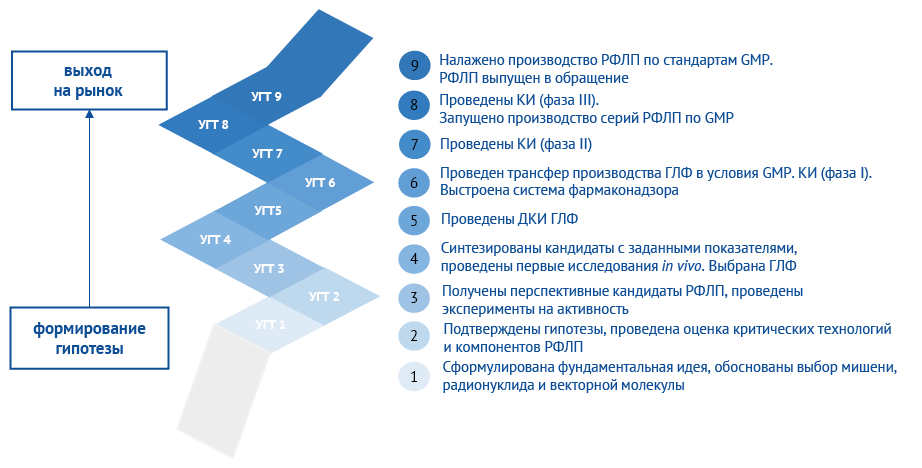
Рисунок подготовлен авторами / The figure is prepared by the authors
Рис. 1. Шкала уровней готовности технологии радиофармацевтических препаратов. РФЛП — радиофармацевтический лекарственный препарат; УГТ — уровень готовности технологии; ГЛФ — готовая лекарственная форма; ДКИ — доклинические исследования; КИ — клинические исследования; GMP — Good Manufacturing Practice, надлежащая производственная практика
Fig. 1. Technology readiness level scale for development of radiopharmaceuticals. RPs, radiopharmaceuticals; TRL, technology readiness level; CTs, clinical trials; GMP, Good Manufacturing Practice
На этапе УГТ 1 (начальный уровень) происходит формирование фундаментальной научной идеи разрабатываемого продукта и обоснование его полезности. При планировании разработки РФЛП необходимо определить заболевания, для лечения которых планируется разработать и внедрить в клиническую практику РФЛП, а также провести аналитический обзор научной литературы на предмет поиска биологической мишени, характерной для выбранных заболеваний; векторной молекулы, тропной к биологической мишени; хелатора, который формирует комплекс между радионуклидом и молекулой-носителем (вектором); диагностического и (или) терапевтического радионуклида для синтеза с конъюгатом. На данном этапе рекомендуется изучить научный опыт по разработке подобных РФЛП, поиск и анализ существующих аналогов разрабатываемого РФЛП или других лекарственных средств на предмет их достоинств и недостатков в диагностике и (или) лечении.
Ключевыми действиями на этом этапе являются детальное планирование и прогнозирование разработки с учетом результатов проведения патентного поиска6 на предмет наличия уже существующих решений, внешних факторов и национальных стандартов, а также существующих технологий производства7.
Учитывая, что каждый последующий этап разработки РФЛП требует еще большего объема затрат, оценку медицинской потребности и подготовку обоснования экономической целесообразности разработки препарата в целом следует выполнить до начала исследований.
На этапе УГТ 2 должны быть выделены целевые области применения продукта, подтверждены обоснованность и эффективность использования предложенной идеи. В случае РФЛП концепция разработки становится более оформленной после дополнительного анализа известных результатов научных исследований для подтверждения гипотезы о тропности векторной молекулы и о биологической мишени, экспериментальной проверки альтернативных концепций, идентификации и оценки критических технологий и компонентов. Как итог, обязательными результатами являются составление целевого профиля качества продукта (Quality Target Product Profile, QTPP), разработка дизайна исследования, проведение скрининга потенциальных соединений in silico (например, с использованием современных цифровых методов и искусственного интеллекта) (см., например, [13][14]), а также подготовка списка потенциальных кандидатов. Важно подчеркнуть, что уже на УГТ 2 необходимо провести комплекс мероприятий по обеспечению правовой охраны результатов интеллектуальной деятельности (РИД), патентные исследования, подать заявки на получение патентов на изобретения и т.д. Недостаточная проработка стратегии правовой охраны РИД, полученных в ходе разработки, — частая ошибка [15], которая впоследствии усложняет процесс коммерциализации и ведет к трудностям при поиске промышленного партнера для вывода продукта на рынок.
УГТ 3 содержит большой блок работ по получению (синтезу) перспективных кандидатов на биологическую мишень и векторную молекулу и подтверждению их тропности экспериментальным путем. На данном уровне выделяют следующие подэтапы:
1) разработка технологии синтеза векторной молекулы;
2) разработка методики контроля качества кандидатов;
3) разработка дизайна предварительных лабораторных исследований кандидатов;
4) подбор лабораторной (in vitro и ex vivo) тест-системы (модели) для проверки активности кандидатов;
5) проведение in vitro исследований эффективности кандидатов на лабораторных моделях — выявление наиболее перспективных пар кандидатов экспериментальным путем;
6) разработка протокола проведения предварительных исследований на лабораторных животных;
7) подбор лабораторных тест-систем для предварительных in vivo исследований фармакокинетики и (или) терапевтической эффективности РФЛП;
8) наработка опытных партий кандидатов по результатам первичного скрининга.
На данном этапе обязательными являются подготовка проекта лабораторного регламента синтеза векторной молекулы и проекта методик контроля качества соединения-кандидата. Важным остается отслеживание и пристальное внимание к обеспечению охраны РИД, своевременная подготовка заявок на выдачу патентов, оформление соглашений с партнерами и соразработчиками с указанием распределения ответственности и прав на РИД.
УГТ 4 включает в себя наработку активной фармацевтической субстанции в лабораторных масштабах, проведение ее ДКИ in vivo и выбор готовой лекарственной формы (ГЛФ). На данном уровне можно выделить 3 блока работ с соответствующим пакетом документов, подтверждающих достижение результатов.
В рамках первого блока осуществляется разработка технологии синтеза направляющей (векторной) молекулы с радионуклидами/радионуклидом, разработка и валидация методики контроля качества РФЛП, наработка опытной партии РФЛП и оценка связываемости и стабильности полученного действующего вещества в покое и при разбавлении.
На втором подэтапе УГТ 4 для РФЛП проводятся предварительные исследования in vivo по фармакокинетике и (или) терапевтической эффективности, по результатам которых может быть скорректирована лабораторная технология синтеза. После актуализации лабораторной технологии можно приступать к разработке нормативной документации на синтезированный РФЛП.
Заключительным подэтапом работ для УГТ 4 является отработка технологии синтеза РФЛП с терапевтическими и (или) диагностическими активностями и выбор ГЛФ («горячий» препарат или лиофилизат для приготовления РФЛП в условиях медицинского учреждения).
Самым важным элементом на УГТ 4 является оценка основных параметров качества кандидата РФЛП. На данный момент утверждены и включены в Государственную фармакопею Российской Федерации XV изд. 32 общие фармакопейные статьи на РФЛП, в том числе статьи на химические предшественники и препараты для позитронно-эмиссионной томографии, а также ряд фармакопейных статей на РФЛП, определяющих специфические параметры качества РФЛП, связанные с радиоактивностью, в частности подлинность по радионуклиду; радионуклидная чистота (РНЧ); радиохимическая чистота (РХЧ); при необходимости объемная (удельная, молярная) активность.
Существуют различные подходы к методам обнаружения и количественного определения радионуклидных, радиохимических и химических примесей [16][17], необходимо также учитывать при разработке препарата влияние примесей на качество визуализации и дозиметрические характеристики.
Согласно накопленному практическому опыту АО «Росатом Наука», на ранних этапах разработки РФЛП (УГТ 1 — УГТ 4) целесообразно уделить особое внимание таким аспектам, как подбор высококвалифицированных специалистов, оснащение научных лабораторий современным оборудованием, разработка и валидация необходимых методик.
Персонал должен быть квалифицированным и уметь надлежащим образом применять асептические приемы в течение всего времени обращения с разрабатываемым РФЛП. Оборудование, применяемое при синтезе РФЛП, контроле качества получаемого продукта, должно быть использовано в соответствии со своим назначением и не приводить к контаминации получаемого продукта. При этом конструкция оборудования, состав расходных материалов должны обеспечивать отсутствие химической активности, кумулятивности и сорбирующих свойств поверхностей, которые могут контактировать с компонентами, используемыми в ходе синтеза и исследований перспективных РФЛП (например, исходные материалы, реактивы, растворители, РФЛП и др.) для исключения влияния на качество конечного продукта. Для контроля качества препаратов-кандидатов следует использовать современные методы и соответствующие методики лабораторных испытаний на РНЧ, объемную активность, РХЧ, показатель рН, содержание химических примесей, изотоничность, стерильность, апирогенность. Лабораторные аналитические методы должны быть пригодны для решения поставленных задач и обладать достаточной чувствительностью, избирательностью, точностью и воспроизводимостью. Таким образом, наличие квалифицированной команды исследователей, высокая оснащенность лаборатории, тщательное следование аналитическим методикам и требованиям к выполнению работ позволят избежать рисков на последующих этапах (УГТ 5 — УГТ 9) неполучения РФЛП надлежащего фармакопейного качества и введения некачественного РФЛП в оборот.
УГТ 5 — этап проведения ДКИ РФЛП. На этом этапе происходит наработка опытных партий РФЛП, проведение исследований на связываемость, стабильность в покое и при разбавлении РФЛП. Параллельно проводят разработку дизайна первичной и вторичной упаковок, выбор партнера-производителя векторной молекулы, отрабатывают технологию синтеза векторной молекулы на производстве (Good Manufacturing Practice, GMP), разрабатывают нормативную документацию на векторную молекулу (спецификация или технические условия) и план ДКИ ГЛФ, проводят ДКИ в условиях надлежащей лабораторной практики (Good Laboratory Practice, GLP) и составляют отчет о ДКИ8.
В рамках реализации данного этапа при разработке своих продуктов АО «Росатом Наука» были выделены следующие ключевые особенности проведения ДКИ для РФЛП, отличающие их от ДКИ других ЛП:
Таким образом, успех доклинического этапа разработки РФЛП зависит от тщательного планирования исследования с учетом радиоактивного распада, проведения ключевых исследований биораспределения и дозиметрии, адаптации протоколов токсикологических исследований к особенностям излучения и выбора релевантных моделей. Итогом проведенных ДКИ должно стать решение о целесообразности перехода к этапу КИ.
УГТ 6 для РФЛП включает в себя трансфер производства ГЛФ в условиях GMP и производство опытно-промышленных серий препарата9, а также построение системы фармаконадзора10 и проведение КИ I фазы11.
Трансфер разрабатываемого РФЛП начинается с определения производителя ГЛФ в виде «горячего препарата» или выбора партнера — производителя «холодного набора» (в случае приготовления РФЛП в клинике), далее происходит трансфер (с масштабированием) технологии производства ГЛФ в промышленные условия (GMP). При этом на УГТ 6 необходимо провести квалификацию производителей или поставщиков сырья и материалов, валидацию аналитических методов контроля качества РФЛП, определить условия хранения сырья и материалов для производства РФЛП, согласовать виды упаковки РФЛП, определить условия хранения и перевозки РФЛП конечному потребителю. Данный подэтап предполагает начало выстраивания системы качества применительно к будущему производству.
В части построения системы фармаконадзора на дорегистрационном этапе необходимо ориентироваться на актуальные регуляторные требования, определяемые Правилами надлежащей практики фармаконадзора ЕАЭС (Good Pharmacovigilance Practice, GVP), утвержденными решением Совета ЕЭК от 03.11.2016 № 87; Федеральный закон от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств» (глава 13) (далее — ФЗ-61); приказом Минздрава России от 07.09.2016 № 682н «Об утверждении формы документа, содержащего результаты мониторинга эффективности и безопасности лекарственного препарата для медицинского применения, проводимого держателем или владельцем регистрационного удостоверения лекарственного препарата либо уполномоченным им юридическим лицом»; приказом Росздравнадзора от 17.06.2024 № 3518 «Об утверждении порядка фармаконадзора лекарственных препаратов для медицинского применения».
В связи с высокой потенциальной токсичностью РФЛП КИ фазы I проводятся не с участием здоровых добровольцев, а с участием пациентов с соответствующим заболеванием. Задачами КИ фазы I являются изучение метаболизма, фармакокинетики, фармакодинамики, дозиметрии, безопасности, лекарственного взаимодействия, предварительная оценка эффективности РФЛП [5]. Подготовка и проведение КИ при разработке РФЛП выполняются согласно обычному алгоритму: формирование и подача пакета документов для получения разрешения на проведение КИ; получение разрешения на проведение КИ; проведение КИ согласно Правилам надлежащей клинической практики (Good Clinical Practice, GCP) и требованиям актуальных регуляторных документов; подготовка и предоставление в уполномоченный орган отчета о проведенных КИ с целью перехода к следующей фазе КИ. Прогностические уровни безопасных диагностических/терапевтических доз, а также предварительные схемы дозирования и введения исследуемого препарата во время проведения I фазы КИ, учитывая специфику РФЛП, устанавливаются на основании результатов ДКИ РФЛП. Переход на УГТ 7 будет осуществляться по завершении КИ фазы I и при наличии отчета о КИ.
На этапе УГТ 7 проводится II фаза КИ12. Изучение эффективности препарата осуществляется на более широкой популяции пациентов по стандартному алгоритму: формирование и подача пакета документов для получения разрешения на проведение КИ; получение разрешения на проведение КИ; проведение КИ согласно GCP; подготовка и предоставление в уполномоченный орган отчета о проведенных КИ с целью перехода к следующей фазе КИ (фаза III). Задачами КИ II фазы являются определение фармакологической эффективности РФЛП, эффективности различных режимов дозирования у особых групп пациентов, оценка безопасности применения препарата.
Также по результатам КИ II фазы может потребоваться корректировка критических показателей качества ГЛФ, аналитических методик ГЛФ, критических параметров процесса и, как следствие, стратегии контроля.
УГТ 8 включает в себя проведение КИ III фазы для подтверждения эффективности и безопасности разработанного РФЛП, производство промышленных серий препарата по стандартам GMP и регистрацию ЛП. КИ III фазы проводится по стандартной процедуре согласно GCP, с аналогичной последовательностью подготовки документов, как и на КИ I и II фазы. По завершении III фазы КИ отчет о проведенных КИ предоставляется в уполномоченный орган. Данный отчет входит в пакет необходимых документов для перехода на следующий — финальный — уровень готовности технологии.
На данном этапе также критически значимыми являются корректировка и актуализация критических параметров процесса производства ГЛФ и стратегии контроля ГЛФ. Необходимо провести валидацию процесса производства и очистки ГЛФ и организовать закладку валидационных серий в достаточном объеме для изучения стабильности препарата.
Заключительным этапом работ на УГТ 8 для других групп ЛП является регистрация лекарственного препарата, но в случае РФЛП возможны разные стратегии последующей реализации продукта (рис. 2), от выбора которых зависит порядок регистрационных процедур: реализация РФЛП с регистрационным удостоверением; применение РФЛП внутри медицинской организации; изготовление и реализация РФЛП через ядерную аптеку.
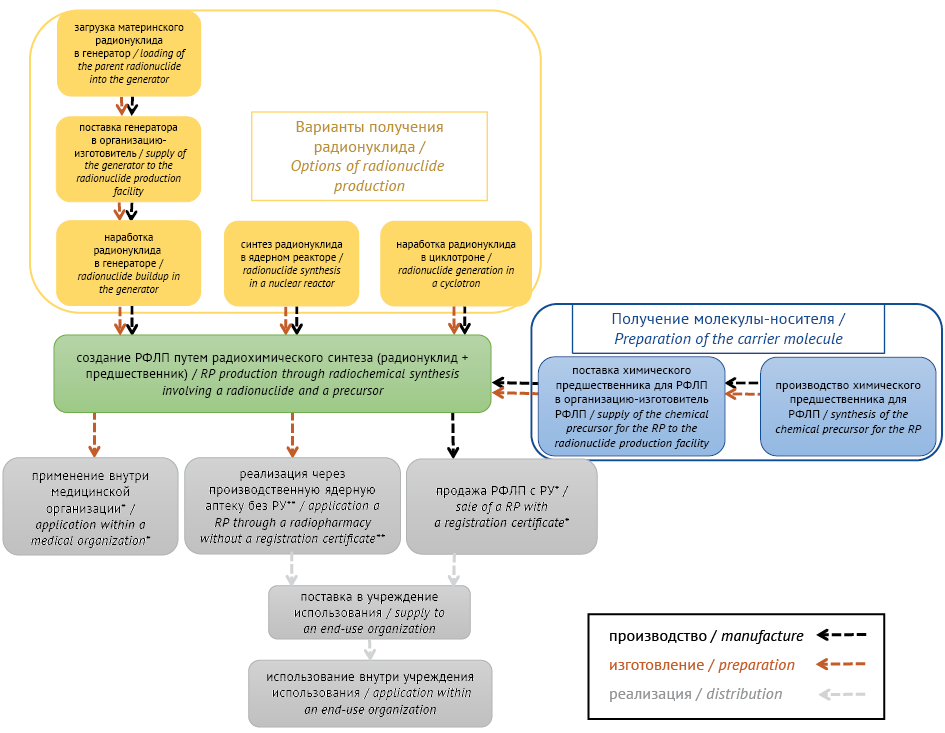
Рисунок подготовлен авторами / The figure is prepared by the authors
Рис. 2. Виды реализации радиофармацевтических препаратов (РФЛП) в Российской Федерации с учетом особенностей изготовления/производства. РУ — регистрационное удостоверение
* РФЛП, которые могут реализовываться путем применения внутри медицинской организации и (или) через продажу с РУ:
1) оригинальные препараты, не находящиеся под охраной патента;
2) оригинальные препараты, если организация является правообладателем.
** РФЛП, которые могут реализовываться через производственную ядерную аптеку, изготавливающую РФЛП:
1) оригинальные препараты, не находящиеся под охраной патента;
2) оригинальные препараты, если организация является правообладателем (обладатель исключительного права на объекты интеллектуальной собственности, в частности патента на изобретение на РФЛП);
3) оригинальные препараты, находящиеся под охраной патента, для разового изготовления без цели получения прибыли (1 пациент — 1 назначение — 1 препарат).
Fig. 2. Marketing options for diagnostic and therapeutic radiopharmaceuticals in the Russian Federation considering manufacturing/production specifics. MA, marketing authorization
* Radiopharmaceuticals that may be distributed within a medical organisation and/or sale with a marketing authorisation:
1) original drugs not protected by patent;
2) original drugs if the organisation is the patent holder.
** Radiopharmaceuticals that may be distributed through a radiopharmacy:
1) original drugs not protected by patent;
2) original drugs, if the organisation is the patent holder (exclusive owner of intellectual property, in particular a patent for a radiopharmaceutical);
3) original drugs protected by patent, for single-use preparation without profit motive (1 patient — 1 prescription — 1 dose).
Согласно п. 4 Решения Совета ЕЭК от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения» (далее — Решение № 78) РФЛП, производимые в промышленных условиях, подлежат регистрации. Также положения об обязательной государственной регистрации РФЛП, производимых в промышленных условиях, содержатся в ст. 13 ФЗ-61. Как правило, данная стратегия применяется для долгоживущих РФЛП или наборов/прекурсоров для изготовления РФЛП. Регистрация данного типа ЛП осуществляется по общему порядку Решения № 78 (раздел V), требования к досье — Приложение № 1 Решения № 78, раздел III.
РФЛП, изготовленные в медицинских организациях по установленным правилам, не подлежат государственной регистрации (п. 5 Решения № 78, ст. 13 ФЗ-61). Порядок обращения РФЛП в медицинских организациях утвержден приказом Минздрава России от 12.11.2020 № 1218н. Изготовление происходит в контролируемых зонах, соответствующих санитарно-эпидемиологическим требованиям и нормативам радиационной безопасности. Медицинская организация должна иметь лицензию на их изготовление, так как это относится к фармацевтической деятельности (п. 33 ст. 4 ФЗ-61, п. 47 ст. 12 Федерального закона от 04.05.2011 № 99-ФЗ «О лицензировании отдельных видов деятельности»).
Также необходимо отметить, что небольшой срок годности РФЛП, изготовляемых в медицинских организациях [18], определяет особенности проведения процедур контроля качества, в частности, в отношении таких препаратов применяется ретроспективный контроль, включающий контроль свойств самого препарата, производственного процесса и исходного сырья.
Изготовление и отпуск ЛП аптечными организациями, включая РФЛП, содержащие один или несколько радионуклидов, используемые для диагностики и терапии, в том числе в позитронно-эмиссионной томографии, регулирует приказ Минздрава России от 22.05.2023 № 249н (часть X). При изготовлении РФЛП используются радионуклидные генераторы, циклотроны и другое специализированное оборудование.
Главным основанием для перехода на следующий уровень готовности технологии будет включение разработанного РФЛП в единый реестр зарегистрированных лекарственных средств ЕАЭС и получение регистрационного удостоверения или разрешения на обращение.
Достижение УГТ 9 (зрелый уровень) соответствует серийному производству РФЛП по стандартам GMP, выпуску РФЛП в обращение, а также включает в себя пострегистрационные исследования/обязательства (при наличии). Из ключевых этапов на УГТ 9 можно выделить промышленное производство в коммерческих объемах РФЛП надлежащего качества, надлежащее функционирование системы фармаконадзора на пострегистрационном этапе, разработку клинических рекомендаций, включение в стандарты медицинской помощи, а также включение ЛП в перечни и минимальный ассортимент лекарственных препаратов13. Основными документами, подтверждающими достижение УГТ 9, являются паспорта качества на промышленные серии препарата, досье на выпущенные в гражданский оборот серии РФЛП, обзоры качества продукции, мастер-файл системы фармаконадзора.
Путь от идеи до вывода на рынок воспроизведенного РФЛП обычно менее длительный и менее затратный по сравнению с разработкой оригинальных ЛП. Основной целью является не поиск нового решения и отбор молекул-кандидатов, а получение продукта, биоэквивалентного референтному ЛП. Первые четыре этапа (УГТ 1 — УГТ 4) в данном случае могут быть проведены гораздо быстрее, так как структура и требования к конечному продукту уже известны. Однако следует отметить, что, поскольку на многие подобные препараты все еще распространяется действие патентов, принадлежащих иностранным владельцам, при разработке воспроизведенных РФЛП часто требуется подбор оригинальной молекулы-мишени, которая будет обладать высокой специфичностью, чувствительностью, эффективностью и безопасностью и при этом быть пригодной для патентной защиты.
Учитывая, что действующее вещество уже изучено и активно применяется в реальной практике, при регистрации воспроизведенного РФЛП нет необходимости в проведении полноценных ДКИ и основных фаз КИ, достаточно получения убедительного доказательства биоэквивалентности воспроизведенного препарата референтному. Объем убедительного доказательства в части наполнения регистрационного досье РФЛП и их прекурсоров представляется в соответствии с требованиями раздела 6 Решения № 78 с учетом правил проведения исследований биоэквивалентности ЛП. Таким образом, особое внимание на УГТ 5 — УГТ 7 необходимо уделить дизайну исследования, зависящему от стратегии последующего введения в обращение.
При разработке РФЛП следует учитывать их многокомпонентную структуру и уделять внимание каждому значимому компоненту состава: векторной молекуле, хелатору, линкеру и радионуклиду. Также следует обратить внимание на предварительное обоснование экономической целесообразности при планировании проекта и обеспечение комплексной правовой охраны разработки. На этапах УГТ 4 — УГТ 5 при оценке основных параметров качества препарата-кандидата и дизайне ДКИ и КИ особое внимание следует уделить параметрам, связанным с радиоактивностью разрабатываемого препарата и выбранной стратегией введения препарата в обращение.
Таким образом, рассмотренная шкала УГТ для РФЛП учитывает основные нюансы разработки РФЛП вне зависимости от выбранной стратегии вывода препарата на рынок и является удобным и гибким инструментом в арсенале исследователей и заказчиков разработки, который позволяет как отслеживать прогресс исследования, так и минимизировать вероятные риски, избежать возможных дорогостоящих ошибок и ускорить вывод РФЛП на рынок, что положительно скажется на доступности современных методов диагностики и терапии для пациентов, а также будет способствовать развитию российской отрасли в целом.
1 https://www.vesti.ru/article/4547948
2 https://tass.ru/ekonomika/24253495
3 http://government.ru/rugovclassifier/926
4 https://tass.ru/obschestvo/23315695
5 Pharmaceutical CGMPs for the 21st century — A risk-based approach. Final report. FDA; 2004.
6 ГОСТ Р 15.011–2024. Интеллектуальная собственность. Патентные исследования. Содержание и порядок проведения.
7 Федеральный закон от 23.08.1996 № 127-ФЗ «О науке и государственной научно-технической политике».
ГОСТ 15.101–2021. Система разработки и постановки продукции на производство. Порядок выполнения научно-исследовательских работ.
ГОСТ 7.32–2017. Межгосударственный стандарт. Система стандартов по информации, библиотечному и издательскому делу. Отчет о научно-исследовательской работе. Структура и правила оформления.
8 Решение Коллегии ЕЭК от 26.11.2019 № 202 «Об утверждении Руководства по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов».
9 Рекомендация Коллегии ЕЭК от 29.01.2019 № 3 «О Руководстве по производству готовых лекарственных форм лекарственных препаратов».
10 Решение Совета ЕЭК от 03.11.2016 № 87 «Об утверждении Правил надлежащей практики фармаконадзора Евразийского экономического союза».
11 Решение Совета ЕЭК от 03.11.2016 № 79 «Об утверждении Правил надлежащей клинической практики Евразийского экономического союза».
Федеральный закон от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств».
12 Решение Совета ЕЭК от 03.11.2016 № 79 «Об утверждении Правил надлежащей клинической практики Евразийского экономического союза».
13 Распоряжение Правительства Российской Федерации от 12.10.2019 № 2406-р «Об утверждении перечня жизненно необходимых и важнейших лекарственных препаратов, а также перечней лекарственных препаратов для медицинского применения и минимального ассортимента лекарственных препаратов, необходимых для оказания медицинской помощи».
1. Kunos CA, Mankoff DA, Schultz MK, et al. Radiopharmaceutical chemistry and drug development — what’s changed? Semin Radiat Oncol. 2021;31(1):3–11. https://doi.org/10.1016/j.semradonc.2020.07.006
2. Zhang S, Wang X, Gao X, et al. Radiopharmaceuticals and their applications in medicine. Signal Transduct Target Ther. 2025;10:1. https://doi.org/10.1038/s41392-024-02041-6
3. Лунев АС, Петросова КА, Терновская КЭ и др. Анализ действующих норм и правил проведения доклинических исследований радиофармацевтических препаратов. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):81–90. https://doi.org/10.30895/1991-2919-2024-14-1-81-90
4. Клементьева ОЕ, Смирнова АВ, Кульбачевская НЮ и др. Доклинические исследования радиофармацевтических лекарственных препаратов: анализ зарубежной и отечественной практики нормативного регулирования (обзор). Регуляторные исследования и экспертиза лекарственных средств. 2024;14(3):251–64. https://doi.org/10.30895/1991-2919-2024-14-3-251-264
5. Горячев ДВ, Лысикова ИВ, Черная АА, Кушнир ДД. Планирование клинических исследований радиофармацевтических лекарственных препаратов: анализ международных рекомендаций и экспертного опыта. Регуляторные исследования и экспертиза лекарственных средств. 2025;15(1):105–20. https://doi.org/10.30895/1991-2919-2025-15-1-105-120
6. Лабушкина АА, Клементьева ОЕ, Кодина ГЕ, Самойлов АС. Разработка методических документов, регламентирующих клинические исследования новых радиофармацевтических лекарственных препаратов. Медицинская радиология и радиационная безопасность. 2023;68(3):71–7. https://doi.org/10.33266/1024-6177-2023-68-3-71-77
7. Косенко ВВ, Трапкова АА, Калмыков СН. Регулирование обращения радиофармацевтических препаратов. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2022;12(4):379–88. https://doi.org/10.30895/1991-2919-2022-12-4-379-388
8. Kapadia R, Shevalkar G, Das U, et al. Introduction to quality by design. In: Jain NK, Neha Bajva, eds. Introduction to quality by design (QbD). Singapore: Springer; 2024. https://doi.org/10.1007/978-981-99-8034-5_1
9. Пятигорский АМ, Бркич ГЭ, Береговых ВВ, Пятигорская НВ. Комплексная оценка технологической готовности инновационного проекта при разработке фармацевтического продукта. Вестник Российской академии медицинских наук. 2023;78(3):234–41. https://doi.org/10.15690/vr-amn8349
10. Gundogdu E, Demir ES, Özgenç E, et al. Applying quality by design principles in the development and preparation of a new radiopharmaceutical: technetium-99m-imatinib mesylate. ACS Omega. 2020;5(10):5297–305. https://doi.org/10.1021/acsomega.9b04327
11. Аксенова ЕИ, Горбатов СЮ, Пивоварова ОА. Определение уровня технологической готовности разработок в медицине на основе методологии TRL. Проблемы социальной гигиены, здравоохранения и истории медицины. 2021;29(спецвыпуск):1395–9. https://doi.org/10.32687/0869-866X-2021-29-s2-1395-1399
12. Кошевенко АС, Деграве ТВ, Буренков ПВ и др. Роль отдельных институтов в реализации достижения фармацевтического суверенитета на примере Центра трансфера медицинских технологий ФГБУ «НЦЭСМП» Минздрава России. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(5):505–12. https://doi.org/10.30895/1991-2919-2024-14-5-505-512
13. Поройков ВВ, Дмитриев АВ, Дружиловский ДС и др. Оценка безопасности фармакологических веществ in silico с применением методов машинного обучения: обзор. Безопасность и риск фармакотерапии. 2023;11(4):372–89. https://doi.org/10.30895/2312-7821-2023-11-4-372-389
14. Sarvepalli S, Vadarevu S. Role of artificial intelligence in cancer drug discovery and development. Cancer Lett. 2025;627:217821. https://doi.org/10.1016/j.canlet.2025.217821
15. Аникеева МЮ, Горбунова ЮА, Пикина НА. Элементы стратегического планирования патентной охраны фармацевтических разработок. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(5):513–20. https://doi.org/10.30895/1991-2919-2024-14-5-513-520
16. Кодина ГЕ, Малышева АО, Ларенков АА, Брускин АБ. Присутствие возможных примесей в радиофармацевтических лекарственных препаратах и методы их определения. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2022;12(3):244–62. https://doi.org/10.30895/1991-2919-2022-12-3-244-262
17. Ларенков АА, Митрофанов ЮА, Рахимов МГ. Особенности и практические аспекты определения радиохимической чистоты рецепторспецифичных препаратов лютеция-177 на примере [177Lu]Lu–PSMA-617. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2022;12(4):455–67. https://doi.org/10.30895/1991-2919-2022-12-4-455-467
18. Шатик СВ, Майстренко ДН, Станжевский АА. Особенности регуляторного статуса радиофармацевтических лекарственных препаратов, изготавливаемых в медицинских организациях. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2022;12(4):389–94. https://doi.org/10.30895/1991-2919-2022-12-4-389-394
Степанова Александра Владимировна.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Тихонова Анна Эдуардовна.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Кошевенко Анастасия Сергеевна - канд. фарм. наук.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Попов Николай Васильевич.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Беланов Константин Юрьевич.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Трапкова Алла Аркадьевна - канд. биол. наук.
Петровский б-р, д. 8, стр. 2, Москва, 127051
Емельянов Владимир Юрьевич.
Кадашевская наб., д. 32/2, стр. 1, Москва, 115035
Суров Сергей Владимирович.
Кадашевская наб., д. 32/2, стр. 1, Москва, 115035
Мансуров Олег Актавианович.
Кадашевская наб., д. 32/2, стр. 1, Москва, 115035
Хрянин Кирилл Александрович.
Кадашевская наб., д. 32/2, стр. 1, Москва, 115035
Казарян Карен Юрьевич.
Кадашевская наб., д. 32/2, стр. 1, Москва, 115035
Степанова А.В., Тихонова А.Э., Кошевенко А.С., Попов Н.В., Беланов К.Ю., Трапкова А.А., Емельянов В.Ю., Суров С.В., Мансуров О.А., Хрянин К.А., Казарян К.Ю. Планирование и управление процессами разработки радиофармацевтических препаратов с применением шкалы уровней готовности технологии. Регуляторные исследования и экспертиза лекарственных средств. 2025;15(4):391-403. https://doi.org/10.30895/1991-2919-2025-15-4-391-403
Stepanova A.V., Tikhonova A.E., Koshevenko A.S., Popov N.V., Belanov K.Yu., Trapkova A.A., Emelyanov V.Yu., Surov S.V., Mansurov O.A., Khryanin K.A., Kazaryan K.Yu. Planning and Management of Radiopharmaceutical Development Processes Using Technology Readiness Level Scale. Regulatory Research and Medicine Evaluation. 2025;15(4):391-403. (In Russ.) https://doi.org/10.30895/1991-2919-2025-15-4-391-403

Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных




























