



Содержание
Перейти к:
 С. Э. Эрдни-Гаряев,
Д. Д. Мамедов,
Д. С. Юрочкин,
Д. Д. Зеликова,
З. М. Голант,
В. С. Фисенко,
И. А. Наркевич
С. Э. Эрдни-Гаряев,
Д. Д. Мамедов,
Д. С. Юрочкин,
Д. Д. Зеликова,
З. М. Голант,
В. С. Фисенко,
И. А. Наркевич https://doi.org/10.30895/1991-2919-2024-590
Перейти к:
ВВЕДЕНИЕ. В настоящее время в Российской Федерации поставлена задача по восстановлению системы производственных аптек как элемента критически значимой инфраструктуры здравоохранения и лекарственного обеспечения населения страны. Для совершенствования регуляторной практики, разработки и внедрения новых передовых подходов при изготовлении и контроле качества лекарственных препаратов (ЛП) аптечного изготовления, необходимо изучить соответствующие положения, посвященные надлежащим практикам изготовления ЛП в зарубежных системах здравоохранения.
ЦЕЛЬ. Анализ организации деятельности и ее нормативного регулирования в сфере изготовления ЛП в Германии для формирования предложений по разработке и внедрению отечественных правил надлежащей практики изготовления и отпуска ЛП.
ОБСУЖДЕНИЕ. В работе представлено продолжение комплексного исследования законодательства Федеративной Республики Германии, которое уточняет особенности действующих положений системы надлежащих практик аптечного изготовления ЛП. Рассмотрены различия подходов при изготовлении ЛП медицинскими и фармацевтическими работниками. Выявлено, что изготовление ЛП врачами реализуется в соответствии с принципами «свободы медицинской практики» и осуществляется фактически без какого-либо регуляторного воздействия. Все аптечные организации являются производственными, что позволяет повысить физическую доступность населения к ЛП. Выявлены особенности системы обеспечения качества, которая основывается на процессном подходе — включает контроль процессов изготовления и описывает методики контроля качества ЛП, в том числе применимость экспресс-методик при изготовлении ЛП. Немецкая концепция надлежащих аптечных практик позволяет осуществлять фармацевтическую разработку экстемпоральных ЛП с использованием доступных инструментов валидации, квалификации, верификации и допускает проведение внутриаптечного контроля качества вне производственной аптеки, изготовившей ЛП. Концепции определения уровней риска позволяют производственным аптекам формировать собственные программы отбора проб. Описаны подходы и требования к организации процессов проведения фармацевтической экспертизы. Установлено, что аптечные организации вправе самостоятельно устанавливать сроки годности на изготавливаемую номенклатуру ЛП.
ВЫВОДЫ. Отдельные практические решения, реализованные в аптечной практике Федеративной Республики Германии, имеют высокую практическую значимость, могут быть использованы при разработке и внедрении российских правил надлежащей практики изготовления и отпуска ЛП, в частности: проведение внутриаптечного контроля качества вне производственной аптеки, изготовившей ЛП; полноценная фармацевтическая разработка технологий изготовления ЛП и методик контроля качества, в том числе экспресс-методик, с использованием доступных производителям лекарственных средств инструментов валидации, квалификации, верификации; самостоятельное установление аптечными организациями сроков годности на изготавливаемую номенклатуру экстемпоральных ЛП; формирование аптечными организациями собственных программ отбора проб.
Эрдни-Гаряев С.Э., Мамедов Д.Д., Юрочкин Д.С., Зеликова Д.Д., Голант З.М., Фисенко В.С., Наркевич И.А. Нормативное правовое регулирование изготовления лекарственных препаратов аптечными организациями на немецком фармацевтическом рынке. Часть 2. Особенности организации деятельности (обзор). Регуляторные исследования и экспертиза лекарственных средств. 2025;15(1):63-81. https://doi.org/10.30895/1991-2919-2024-590
Erdni-Garyaev S.E., Mamedov D.D., Yurochkin D.S., Zelikova D.D., Golant Z.M., Fisenko V.S., Narkevich I.A. Pharmacy Compounding Regulation in the German Pharmaceutical Market. Part 2. Organisational Features (Review). Regulatory Research and Medicine Evaluation. 2025;15(1):63-81. (In Russ.) https://doi.org/10.30895/1991-2919-2024-590
В настоящее время в Российской Федерации поставлена задача по восстановлению системы производственных аптек как элемента критически значимой инфраструктуры здравоохранения и лекарственного обеспечения населения страны.
В первой части исследования [1] были раскрыты основные императивы законодательства Федеративной Республики Германия (далее — Германия, ФРГ) об обращении лекарственных средств (ЛС), продемонстрированы различия российского и немецкого законодательства, в том числе относительно лингвистических единиц фармацевтических терминов и определений, а также их значений. Так, в нормативном поле Германии фармацевтические субстанции (далее — АФС) являются отдельной сущностью и не входят в дефиницию ЛС, в свою очередь, последние представлены в виде составных ЛС (далее — РЭЛП), а также готовых лекарственных препаратов, состоящих из зарегистрированных лекарственных препаратов (готовых лекарственных форм; далее — ГЛФ) и внутриаптечной заготовки (ВАЗ). Описанное значительно отличается от понятийного аппарата Федерального закона Российской Федерации от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств» (ФЗ-61), где изготавливаемые лекарственные препараты (далее — экстемпоральные лекарственные препараты, ЭЛП) являются лекарственными препаратами (ЛП) и могут быть изготовлены индивидуально для пациента или группы пациентов по факту поступления рецепта на ЛП, а также требования медицинской организации (МО), или предварительно изготовлены в виде ВАЗ. Настоящая статья продолжает сравнительное исследование немецкого права в сфере отношений, связанных с изготовлением ЛС в аптечных организациях.
Цель работы — анализ организации деятельности и ее нормативного регулирования в сфере изготовления лекарственных препаратов в Германии для формирования предложений по разработке и внедрению отечественных правил надлежащей практики изготовления и отпуска лекарственных препаратов.
Задачи исследования:
В работе проведен анализ нормативных правовых документов и баз данных Германии (Gesetze im Internet1), судебных решений, опубликованных в базе Dejure2, статистических данных, доступных в открытых источниках. Поиск осуществлялся за период 1949 (год принятия основного закона ФРГ) — 2023 гг. по следующим поисковым словам: «Wirkstoffe», «Rekonstitution eines Fertigarzneimittels», «Apotheke», «Anerkannte pharmazeutische Regeln», «Deutschen Arzneibuch», «freiverkäufliche Arzneimittel», «Rahmen des üblichen Apothekenbetriebes», «Standardzulassung», «Deutscher Arzneimittel-Codex / Neues Rezeptur-Formularium», «Arzneimittelpreisverordnung».
Необходимо упомянуть, что в немецком законодательстве для обозначения изготовления ЛП и производства ЛС используется один и тот же термин (нем. Herstellen), и далее по тексту предложено использовать такие понятия, как «изготовление ЛП», «производство ЛС», «производство (изготовление)» в зависимости от контекста описываемых положений немецкого законодательства. Также во избежание двойного толкования определений специализации фармацевтических работников необходимо отметить, что в большей части стран мира фармацевт — это специалист с высшим фармацевтическим образованием (в Российской Федерации — провизор), а помощник фармацевта — специалист со средним фармацевтическим образованием (в Российской Федерации — фармацевт). По всему тексту публикации под дефиницией «фармацевт» следует понимать «провизор».
Авторы подчеркивают, что данная работа выполнена в виде двух взаимосвязанных статей и рекомендуют комплексно рассматривать изучение вопросов изготовления ЛП аптечными организациями в рамках немецкого законодательства. Авторы статьи настоятельно рекомендуют читателю ознакомиться с первой частью проведенных изысканий [1], без чего будет достаточно проблематично понять содержание настоящей публикации, которая описывает систему надлежащих аптечных практик действующей на территории Германии.
В целом исследование является продолжением цикла работ авторов, которые посвящены формированию единой гармонизированной системы нормативного правового регулирования в области обращения ЛП, изготавливаемых аптечными организациями (АО) в Российской Федерации (см., например, [1–3]).
Врач в Германии вправе осуществлять медицинскую деятельность в качестве индивидуального предпринимателя [1]3. Согласно Федеральным медицинским правилам от 02.10.19614 врач служит здоровью человека и нации в целом, одновременно медицинская практика по своей природе — «свободная профессия». Последнее выразилось в том, что более чем 30 лет положения действующего на территории Германии закона «О лекарственных средствах» от 24.08.1976 (далее — Закон Германии)5 не распространялись на деятельность врачей по изготовлению ЛС.
В 1997 г. Федеральное министерство здравоохранения Германии (далее — Минздрав Германии) запретило под угрозой уголовного преследования использовать вещества, полученные из органов и тканей организма человека или животного при производстве (изготовлении) ЛС в парентеральных лекарственных формах (ЛФ). Четверо врачей, посчитав такое решение ограничивающим их в правах, подали в суд жалобу. Федеральный конституционный суд Германии в 2000 г. в своем решении — 1 BvR 420/97 (далее — Решение ФКС)6 установил, что изготовление ЛС врачами является «традиционной областью их деятельности» и «существенной частью свободы медицинской практики». Введенный запрет, кроме того, противоречил ч. 19 ст. 74 Конституции Германии7, по смыслу которой под надзор федеральных органов государственной власти не подпадают ЛС, изготавливаемые врачами, поскольку их обращение не является отпуском или реализацией (продажей) ЛС.
Решение ФКС привело к тому, что в 2002 г. в Закон Германии были внесены поправки, согласно которым деятельность по изготовлению ЛС врачами для своих пациентов признавалась не регулируемой законодательством об обращении ЛС. Позже Рабочая группа Бундестага и Бундесрата по модернизации федерального государственного устройства (нем. Kommission von Bundestag und Bundesrat zur Modernisierungder bundesstaatlichen Ordnung) представила доклад8, содержащий оценку полномочий федеральных органов государственной власти в сфере обращения ЛС, согласно которой ч. 19 ст. 74 Конституции Германии противоречит принципам охраны здоровья граждан. В 2003 г. в результате деятельности совместной комиссии Бундестага и Бундесрата по модернизации федерального государственного устройства было принято решение о расширении полномочий федеральных органов государственной власти в отношении изготавливаемых врачами ЛС. Соответствующие поправки были внесены в Конституцию Германии в 2006 г., переходный период изменений занял 3 года и завершился корректировкой Закона Германии в 2009 г.
На сегодняшний день основными документами, регулирующими деятельность АО по изготовлению ЛС, являются закон «Об аптечных организациях» от 20.08.1960 (далее — Закон об аптеках)9 и постановление Минздрава Германии «О работе аптечных организаций» от 09.02.1987 (далее — Постановление НАП)10, однако в отношении изготовления ЛС врачами установленные нормативные правовые требования отсутствуют.
Врач, осуществляющий изготовление ЛС, обязан уведомить об этом региональный орган исполнительной власти (ч. 1–2 ст. 67 Закона Германии), указав наименование и состав ЛС. Содержание и форма такого уведомления без получения лицензии на производство ЛС не унифицированы и различаются в разных регионах. Основная информация, которая должна быть в указанном уведомлении, помимо наименований и состава ЛС включает: ФИО врача, контактную информацию; адрес осуществления деятельности по изготовлению ЛС; заявление врача, что все ЛС изготавливаются исключительно под его прямую профессиональную ответственность с целью личного назначения и применения у конкретного пациента. Некоторые региональные органы исполнительной власти дополнительно запрашивают информацию: о квалификации сотрудников, задействованных при изготовлении ЛС11; об используемых АФС и их поставщиках; о производственном оборудовании; о соблюдении положений признанных фармацевтических правил [1].
Сроки предоставления ответа от компетентного регионального органа исполнительной власти и размер пошлины варьируются в зависимости от сложности состава, лекарственной формы ЛС, а также региона Германии.
Согласно Постановлению НАП все АО Германии, за исключением аптек-филиалов [1], должны иметь техническую возможность самостоятельно изготавливать ЛС, а также проводить контроль качества АФС и вспомогательных веществ (далее — ВВ).
В ст. 1а Постановления НАП установлены следующие термины и определения, касающиеся изготовления ЛС:
В Германии при изготовлении ЛС используются в том числе АФС и ГЛФ. Прямой законодательный запрет на изготовление ГЛФ отсутствует, что подтверждается арбитражной практикой. Например, согласно решению Высшего земельного суда Гамбурга от 18.12.201512 изготовление ЛС зависит исключительно от конкретного рецепта для пациента, при этом изготовление ЛС возможно из ГЛФ. Подобная интерпретация «привилегии ЭЛП» (нем. Rezepturprivileg) вытекает из ч. 1 ст. 3 Директивы 2001/83/ЕС13 и связанного с ней решения Европейского суда от 16.07.201514, согласно решению Окружного суда Гамбурга от 04.02.202115.
Система обеспечения качества экстемпоральных лекарственных препаратов. В соответствии с пояснительной запиской к четвертым поправкам в Постановление НАП от 3 февраля 2012 г.16 (далее — 4-е поправки Постановления НАП) вносимые ими изменения представляют собой значительное обновление немецкой концепции изготовления ЛС, гармонизированное с современными представлениям о данном виде деятельности и разработанное на основании резолюции Комитета министров Совета Европы «О требованиях к обеспечению качества и безопасности лекарственных средств, изготавливаемых в аптеках для особых нужд пациентов» (далее — Резолюция ResAP) [2].
Указанными выше поправками Постановления НАП установлены требования по наличию системы менеджмента качества, разработанной и утвержденной в каждой АО. По смыслу п. 2а Постановления НАП заведующий аптекой несет ответственность за качество изготовленных в аптеке ЛС, а также за разработку системы менеджмента качества (нем. Qualitätsmanagementsystem; далее — СМК). Для функционирования СМК должны быть сформированы стандартные операционные процедуры, обеспечивающие изготовление, контроль качества и хранение ЛС в соответствии с действующими стандартами. Заведующий аптекой отвечает за проведение самоинспекций, кроме того, АО обязана не реже 1 раза в год проводить контроль качества ЭЛП в аккредитованных испытательных лабораториях (центрах). В Германии развита система добровольной сертификации СМК аптечных организаций региональными палатами фармацевтов [1] согласно стандарту ISO 900117. При этом Федеральная палата фармацевтов определила единые требования к органу по сертификации, процессу сертификации и аудиторам. На сегодняшний день единым требованиям соответствуют только 7 из 17 региональных палат фармацевтов.
СМК — непрерывный процесс, охватывающий все сферы деятельности АО, служит основой для поддержания и постоянного улучшения качества оказываемых фармацевтических услуг. Исходя из положений пп. 4а, 7 и 8 Постановления НАП, требований немецкого национального формуляра (нем. Deutscher Arzneimittel-Codex / Neues Rezeptur-Formularium; далее — Формуляр DAC/NRF)18, заведующий аптекой утверждает следующие виды инструкций, входящие в СМК аптечной организации (включая, но не ограничиваясь): инструкцию по изготовлению (нем. Herstellungsanweisung); инструкцию по санитарному режиму (нем. Hygieneplan); инструкцию по испытаниям (нем. Prüfanweisung); систему управления рисками при контроле качества ВАЗ (нем. Prüfung von Defekturarzneimitteln nach Risikomanagement).
В силу п. 4 Постановления НАП производственные помещения АО должны соответствовать определенному типу, размеру, расположению, иметь оборудование, необходимое для изготовления ЛС, обеспечивать надлежащую работу производственных аптек: подготовительные работы, изготовление, контроль качества, упаковку, хранение, а также транспортировку (доставку) ЛС. Производственные помещения должны быть отделены от других помещений аптеки; обеспечены необходимой системой кондиционирования, вентиляции, отвечать требованиям освещенности рабочих мест; быть контролируемыми, то есть подвергаться мониторингу окружающей среды.
Минимальным набором помещений АО являются торговый зал; ассистентская, оборудованная вытяжным шкафом; помещения для хранения исходного сырья и ЛС; комнаты ночного дежурства. Общая площадь указанных помещений должна составлять не менее 110 м². При расчете площади производственной аптеки не учитываются помещения для индивидуальной переупаковки ЛС, а также чистые помещения для изготовления стерильных ЛС.
Для изготовления нестерильных ЛС должна быть выделена отдельная рабочая зона с трехсторонними перегородками, за исключением случаев, когда рабочая зона находится непосредственно в ассистентской. Стены и поверхности, а также пол должны легко очищаться, чтобы свести к минимуму риск загрязнения окружающей среды изготавливаемыми ЛС. Аптеки-филиалы должны состоять как минимум из торгового зала, помещения для хранения ЛС и комнаты для ночного дежурства.
Каждой производственной аптеке необходимо в обязательном порядке иметь оборудование, предназначенное для изготовления суспензий, растворов и эмульсий, в том числе в виде капель (глазных, ушных и назальных), мазей, кремов, гелей, паст, капсул, порошков, суппозиториев.
В аптечной организации должна быть обеспечена возможность изготовления стерильных ЛС (за исключением парентеральных ЛФ). Если в производственной аптеке отсутствует оборудование для получения воды для инъекций, то изготовление ЛС осуществляется из готовой лекарственной формы воды для инъекций, которая в достаточном количестве должна храниться в материальной комнате.
До 2012 г. Постановление НАП включало в себя Приложение № 1, в котором указывался перечень необходимого оборудования, средств измерений, лабораторной посуды и титрованных растворов. Согласно пояснительной записке к 4-м поправкам в Постановление НАП, Приложение № 1 было отменено в рамках устранения административных барьеров при организации деятельности по изготовлению ЛС, поскольку не представляется возможным создать какие-либо избыточные и достаточные списки чего-либо в рамках производственной деятельности АО ввиду высокого уровня вариативности технологических процессов и номенклатуры ЭЛП. При этом заведующий аптекой по собственному решению может приобрести современное оборудование, соответствующее текущему уровню развития фармацевтической науки, вместо ограничительного перечня, ранее установленного Приложением № 1 к Постановлению НАП.
Исходя из положений п. 4а Постановления НАП изготовление ЛС осуществляется в условиях микробиологического контроля как самих ЭЛП, так и окружающей среды. Заведующий аптекой должен составить инструкцию по санитарному режиму, в которой отображены соответствующие санитарно-эпидемиологические меры в отношении персонала, процессов и производственных помещений. Степень и уровень микробиологического контроля должны быть сформированы исходя из требований, установленных Европейской фармакопеей. Инструкция по санитарному режиму не унифицирована для всех АО и разрабатывается производственной аптекой в рамках СМК самостоятельно.
Согласно п. 6 Постановления НАП ЭЛП должны соответствовать требованиям качества, утвержденным нормативными правовыми документами. Альтернативные методики контроля качества, отличные от тех, которые описаны в Немецкой или Европейской фармакопеях (далее — Фармакопея), также могут быть использованы при условии их валидации. Контроль качества ЭЛП может проводиться вне АО под ответственность заведующего аптекой: в компаниях, имеющих лицензию на производство ЛС на территории Германии; в компаниях, имеющих лицензию на производство ЛС на территории государств — членов Европейского союза; в других производственных аптеках, обладающих соответствующей технической возможностью проведения необходимых видов контроля качества; экспертами, имеющими немецкую лицензию на фармацевтическую практику или свидетельство о сдаче экзамена после завершения не менее 4 лет обучения в университете в области фармации, химии, фармацевтической химии и технологии, биологии, медицины человека, а также обладающих практическим опытом работы в сфере контроля качества ЛС не менее 2 лет.
Предприятия или эксперты, ответственные за проведение контроля качества, должны подтвердить, что ЭЛП были проверены согласно признанным фармацевтическим правилам, указав серию, дату и результаты испытаний, в результате чего составляется сертификат испытания (нем. Prüfzertifikat). Непосредственно в АО должна быть как минимум установлена подлинность ЛС и сделаны соответствующие записи о проведенных испытаниях.
В соответствии с п. 7 Постановления НАП РЭЛП должны быть изготовлены по заранее составленным инструкциям для конкретного РЭЛП, в которых указывается технология изготовления и используемое оборудование, методики и виды контроля качества, упаковка и маркировка РЭЛП. Допускается использовать инструкции по изготовлению, разработанные в сторонних организациях после проведения процедуры их верификации в конкретной АО.
Фармацевтическая экспертиза. Перед изготовлением РЭЛП фармацевт должен провести фармацевтическую экспертизу (нем. Plausibilitätsprüfung) рецепта. Данные о фармацевтической экспертизе рецепта и процессе изготовления РЭЛП должны быть задокументированы фармацевтом в протоколе процесса изготовления (нем. Herstellungsprotokoll, аналогично российскому паспорту письменного контроля, далее — ППК), в котором по каждому РЭЛП заполняются: наименование и количество исходного сырья с обозначением их серий или номеров испытаний, параметры процессов изготовления (например, скорость и время перемешивания смеси), результаты внутриаптечного контроля, ФИО пациента и врача (при наличии рецепта), ФИО пациента (при наличии требования физического лица [1]), ФИО фармацевта, изготовившего РЭЛП.
Фармацевтическая экспертиза рецепта проводится, когда ЛС изготавливается впервые или были опубликованы новые научные данные относительно изготавливаемой номенклатуры ЛС (стабильность, токсичность, несовместимость и др.). Фармацевтическую экспертизу часто встречающихся рецептов не требуется проводить перед каждым отдельным изготовлением, поскольку соответствующая процедура была проведена ранее, а данные по ней указаны в ППК. При этом рекомендуется архивировать документацию по фармацевтической экспертизе в соответствии с действующей СМК аптечной организации, а также присваивать каждой рецептуре краткое наименование для обеспечения быстрого доступа к документации.
Основными элементами фармацевтической экспертизы являются анализы:
В результате проведенных действий делается заключение о возможности изготовления РЭЛП. Фармацевт должен проконсультироваться с врачом, выписавшим рецепт, если во время проведения фармацевтической экспертизы возникли какие-либо несоответствия по вопросам, перечисленным выше. Замена фармацевтом АФС осуществляется исключительно по согласованию с врачом, ограничение не распространяется на ВВ. Результат консультации с врачом и вытекающие из этого изменения должны быть задокументированы в письменной форме. ППК заверяется подписью фармацевта.
Объем фармацевтической экспертизы может быть уменьшен при изготовлении стандартизированных рецептур Формуляра DAC/NRF, поскольку в нем могут быть данные о совместимости исходного сырья, стабильности ЭЛП, сроков годности [4].
Установление сроков годности. По итогам фармацевтической экспертизы устанавливается срок годности (нем. Haltbarkeit) на ЭЛП. В Германии, согласно Формуляру DAC/NRF, различают следующие понятия по отношению к сроку годности исходного сырья и ЛС:
В ходе проверки подлинности исходного сырья АО устанавливает срок использования, поскольку производитель исходного сырья, как правило, указывает срок действия или дату окончания срока годности для запечатанной упаковки. В связи с тем что при проверке подлинности производственной аптекой целостность упаковки нарушается, срок использования никогда не может быть дольше указанного производителем срока действия и может быть сокращен в случае, если вскрытая упаковка исходного сырья характеризуется коротким сроком действия. Формуляр DAC/NRF предоставляет табличные значения для сроков использования некоторых видов исходного сырья (табл. 1. «Пример установления срока использования исходного сырья в аптечной организации Германии». Опубликована на сайте журнала19).
Согласно п. 14 Постановления НАП, ЭЛП должны быть промаркированы с указанием срока использования и, при необходимости, срока использования после вскрытия упаковки. В формуляре DAC/NRF в отношении РЭЛП используется термин «срок действия» вместо термина «срок использования», указанного в тексте Постановления НАП, что позволяет однозначно разделить исходное сырье. Указание срока действия после вскрытия упаковки соответствует сроку использования после вскрытия РЭЛП. Бóльшая часть РЭЛП представляет собой многодозовые контейнеры, в таких случаях срок действия теряет свою значимость в отличие от срока использования после вскрытия.
Срок действия и срок использования после вскрытия для стандартизованных рецептур указан в Формуляре DAC/NRF; кроме того, в нем также представлены справочные данные и рекомендуемые сроки использования после вскрытия для ЭЛП, характеризующихся физико-химической стабильностью при их хранении в многодозовых упаковках. При проведении фармацевтической экспертизы должны проводиться дифференциация параметров ЛФ, первичной упаковки, риска микробной контаминации, в соответствии с чем также определяются сроки годности. Таким образом, АО вправе самостоятельно установить срок годности ЭЛП без проведения исследований на стабильность на основании научных данных о стабильности отдельных компонентов ЛС, их физико-химических свойств и взаимодействий, а также проводить собственные исследования на стабильность конкретных рецептур.
Немаловажным фактором, оказывающим влияние на срок годности20 ЛС, является наличие воды: кремы, гидрогели или водные растворы более восприимчивы к микробной контаминации, чем липофильные ЛФ. Согласно Формуляру DAC/NRF, ЛС, не содержащие консервантов, то есть со сроком годности менее 28 сут, не должны изготавливаться как ВАЗ. Максимально возможные сроки годности ЭЛП в различных ЛФ представлены в таблице 2 «Максимальные сроки годности некоторых лекарственных форм экстемпоральных лекарственных препаратов в Германии», опубликованной на сайте журнала21.
Согласно требованиям Фармакопеи исходное сырье должно соответствовать качественным и количественным характеристикам, указанным в спецификации. То есть по состоянию на последний день срока годности количественное содержание веществ должно составлять ≈100% (показатель качества, уменьшающегося во времени; далее — критерий приемлемости). Для ГЛФ критерий приемлемости составляет 95% согласно Фармакопее, в то же время для ЭЛП критерий приемлемости составляет 90% исходя из требований Формуляра DAC/NRF. Таким образом, в случаях, когда используется ГЛФ в качестве исходного сырья при изготовлении ЭЛП, необходимо учитывать остаточный срок годности ГЛФ. Например, если используемая ГЛФ имеет срок использования 4 недели после вскрытия, то срок годности ЭЛП не должен превышать 4 недели.
Процессы изготовления и контроля качества экстемпоральных лекарственных препаратов. Проведение полного химического контроля может не осуществляться в том случае, если качество РЭЛП гарантируется технологическим процессом изготовления (нем. Herstellungsverfahren), органолептическим контролем ЛС, а также в случаях, если это предусмотрено результатами внутрипроцессного контроля (нем. Inprozesskontrollen) — набора испытаний, которые осуществляются в критических точках процесса изготовления ЛС и гарантируют получение ЭЛП надлежащего качества. Кроме того, внутрипроцессный контроль обязателен при изготовлении стерильных ЭЛП (как минимум, мониторинг окружающей среды). Другими примерами внутрипроцессного контроля являются бесконтактное измерение температуры с помощью инфракрасного лазерного термометра; визуальный контроль однородности структуры и физической стабильности; контроль цвета, запаха, значения pH; контроль плотности, насыпной плотности; проверка целостности фильтра путем испытания на определение насыщенности («точка пузырька») при стерилизующей фильтрации; контроль в процессе изготовления, в том числе для ЛС, изготовленных с использованием автоматизированных систем перемешивания.
Изготовление ЭЛП осуществляется согласно «принципу четырех глаз» (нем. 4-Augen-Prinzip), сопоставимым с российским правилом «изготовления под наблюдением провизора-технолога или провизора-аналитика в случае отсутствия методик контроля качества»22, однако в данном контексте речь идет не только про физическое наблюдение за изготовлением конкретного ЛС. При этом, одновременное изготовление ЛС в АО на различных производственных участках требует регулярного контроля вне зависимости от видов ЛФ. Таким образом, «принцип четырех глаз» в первую очередь подразумевает под собой надлежащий уровень контроля за процессами изготовления ЛС.
На рисунке 123 представлена блок-схема принятия решения по изготовлению РЭЛП.
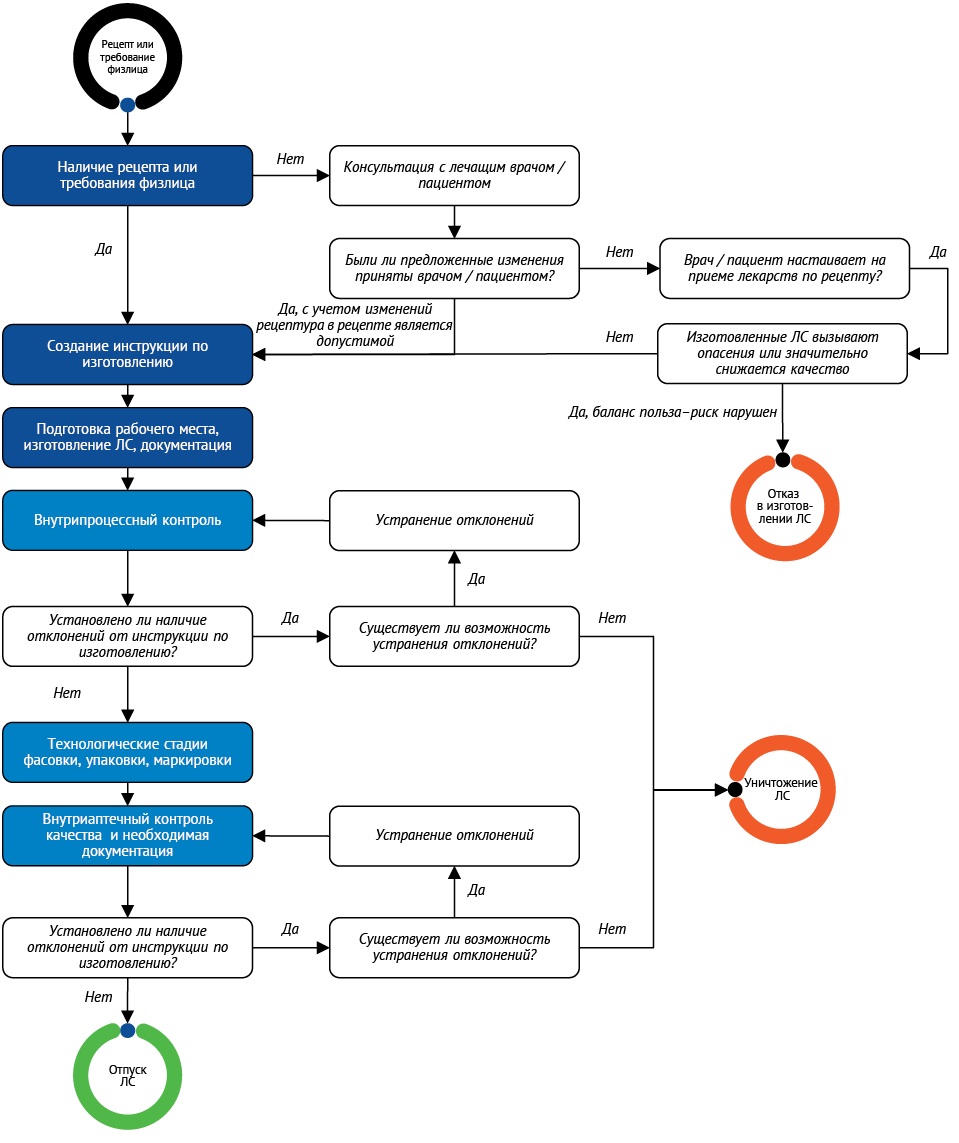
Рисунок подготовлен авторами / The figure is prepared by the authors
Рис. 1. Блок-схема изготовления составных лекарственных препаратов. ЛС — лекарственное средство
Fig. 1. Flowchart for compounding extemporaneous formulations23
Процесс изготовления и контроля качества внутриаптечной заготовки. Согласно п. 8 Постановления НАП, ВАЗ, аналогично РЭЛП, изготовляется в соответствии с предварительно (заранее) разработанными инструкциями по изготовлению. В отличие от РЭЛП, ВАЗ считается готовым лекарственным препаратом, поэтому в инструкции по изготовлению также должна быть указана информация, каким образом препарат вводится в гражданский оборот (основание для изготовления и отпуска ВАЗ: рецепт, требование физического лица, договор поставки или «стандартное разрешение» [1]). При изготовлении ВАЗ информация также фиксируется в ППК, в котором должны быть указаны дата изготовления и номер серии; наименование и количество исходного сырья с обозначением серии или номера испытания; результаты внутриаптечного контроля; параметры процессов изготовления; количество изготовленных единиц или разведенных (восстановленных) ГЛФ [2]; срок годности; подпись лица, изготовившего ВАЗ.
Для внутриаптечного контроля ВАЗ разрабатывается соответствующая инструкция по испытаниям, содержащая подробную информацию об отборе проб, методиках проводимых исследований и др. По итогам проверки контроля качества ВАЗ оформляется протокол испытаний (нем. Prüfprotokoll), который включает в себя дату проведения испытаний, их результаты, подпись и ФИО лица, ответственного за проведение контроля качества, подпись и ФИО заведующего аптекой. Согласно 4-м поправкам Постановления НАП, протокол испытания — фундаментальная часть признанных фармацевтических правил, в том числе правил Надлежащей производственной практики (Good Manufacturing Practice, GMP), при этом под внутриаптечным контролем ВАЗ не следует понимать полное аналитическое тестирование (например, согласно требованиям Фармакопеи, включая определение примесей, остаточных растворителей и др.). Текст Постановления НАП не содержит конкретных указаний относительно объема и видов испытаний, в отличие от подхода, реализованного в Фармакопее США [2][3]. Следует подчеркнуть, что в контексте аптечного изготовления ЛС необходимо учитывать факторы, существенно разграничивающие ЭЛП и ГЛФ. Для ЭЛП характерно единичное изготовление ЛС; меньший размер серии ВАЗ; изготовление ЭЛП исключительно фармацевтическими работниками (в отличие от ГЛФ, где подобное требование отсутствует); отсутствие государственной регистрации ЭЛП, в связи с чем врач, назначивший ЭЛП, а также фармацевтические работники, задействованные при изготовлении ЛС, несут персональную ответственность перед пациентом в пределах своей компетенции.
Сочетание упомянутых факторов обосновывает сокращение объема контролируемых упаковок ЭЛП и видов контроля качества до необходимого минимума. Решение о том, что включает этот минимум, принимает ответственный за изготовление фармацевт или заведующий аптекой. Такие решения должны осуществляться на основе оценки риска при изготовлении ЛС и документироваться для обеспечения прослеживаемости согласно требованиям монографии 2619 «Фармацевтические препараты» Европейской фармакопеи. Оценка уровня риска является предварительным этапом, на основе которого определяются оптимальные методы испытаний ВАЗ до степени, при которой достигается экономическая целесообразность изготовления ЛС.
Таким образом, заведующий аптекой перед изготовлением первой серии ВАЗ должен заранее составить инструкцию по испытаниям, в которой будет отображена оценка уровня риска. При этом заведующий аптекой может самостоятельно разработать систему оценки риска, которая будет определять влияние различных параметров на ВАЗ (например, качество АФС, ВВ и упаковки, процесса изготовления, объем и значимость испытаний, стабильность препарата) или использовать разработанные, нормативно закрепленные концепции (методы) оценки риска. Базовое описание концепции оценки уровня риска при изготовлении ЛС изложено в Резолюции ResAP [2], в которой предлагается типовая модель оценки с выделением ЭЛП высокого или низкого уровней риска. В таблице 3 «Критерии принятия решения при оценке риска согласно Резолюции ResAP» (опубликована на сайте журнала)24 приведены критерии для принятия решения при оценке уровня риска ЭЛП. Каждому критерию присваивают коэффициенты от 1 до 5. Число, полученное в результате умножения этих коэффициентов, указывает на уровень требуемой системы обеспечения качества при изготовлении ЭЛП: если оно превышает 100, ЭЛП относят к категории «препаратов с высоким уровнем риска», если число равно или меньше 100, ЭЛП относят к «препаратам с низким уровнем риска».
В зависимости от результатов оценки риска ЭЛП должны быть задокументированы:
На основании Резолюции ResAP в Германии разработаны две концепции оценки уровней риска изготавливаемой номенклатуры ЭЛП: от Рабочей группы фармацевтов Германии (нем. Arbeitsgemeinschaft der Pharmazieräte Deutschlands; далее — Ассоциация APD) и Формуляра DAC/NRF.
Ассоциация APD в 2013 году выпустила резолюцию по внутриаптечному контролю ВАЗ (далее — Резолюция APD 2013)25, согласно которой только органолептических испытаний ВАЗ, как при изготовлении РЭЛП, недостаточно. Выбор методов испытаний и их объем зависят от потенциального уровня риска, связанного с изготовлением ЛС. Для этого в производственной аптеке должна быть создана система управления рисками, в которой заведующий аптекой классифицирует ВАЗ по одной из четырех категорий уровня риска. При этом он должен оценивать риски в отношении дозировки, токсикологического потенциала, терапевтического диапазона и силы действия, способа применения и ЛФ, безопасности процессов изготовления, размеров серии и частоты ее изготовления.
На основании разработанной системы управления рисками заведующий аптекой индивидуально и самостоятельно определяет методы и объем аналитических испытаний с использованием ступенчатой модели (табл. 4)26.
Таблица 4. Категории рисков и примеры внутриаптечного контроля
Table 4. Risk categories and internal quality control examples26
|
Категория риска |
Описание |
Внутриаптечный контроль |
|
Низкий |
Отсутствуют измеримые критерии риска. Сборы с низкоактивными веществами, ЛФ для наружного применения с низкоактивными веществами. Отсутствует какой-либо риск для здоровья пациента |
Визуальная проверка однородности, определение внешнего вида и цвета, определение размера частиц, измерение pH, определение показателя преломления |
|
Средний |
Выявлен один из критериев риска или собственная оценка аптечной организации показывает, что необходимы дополнительные испытания. ЛФ для наружного применения: мази, кремы, лосьоны, гели с АФС средней активности (например, глюкокортикоиды 2 и 3 класса). Существует потенциальный риск для здоровья пациента |
Измерение pH, определение плотности, объема или веса капель (стандартный счетчик капель), показателя преломления, дисперсности (микроскопия), размера частиц, испытание вытягиванием на стеклянной пластинке, пенетрометрия (консистенция), сухой остаток, экстензометрия, определение коллоидной стабильности. Если целевые значения для этих тестов не могут быть найдены в литературе, они могут быть установлены в аптечной организации |
|
Высокий |
Выявлено несколько критериев риска. ЛФ для перорального применения, суппозитории, глазные капли, растворы для внутриполостного полоскания. Существует значительный потенциальный риск для пациента |
Все виды испытаний для ЛС «среднего риска», а также полуколичественные и количественные аналитические методы, такие как сравнение цвета и помутнения, оценка размера пятна на хроматографической пластине, однородность веса (весовой тест) |
|
Очень высокий |
Выявлены все факторы риска. Парентеральные ЛФ, цитостатические ЛС. Существует значительный потенциальный риск для пациента |
Необходимо соблюдать требования п. 35 Постановления Минздрава Германии «О работе аптечных организаций»27, «Параметрический выпуск» (нем. Parametrische Freigabe), основанный на валидации процессов изготовления ЛС и мониторинге процесса посредством ежемесячного тестирования на частицы и микробы (помещение, персонал), ежемесячное изготовление модельного ЛС, проверка каждой серии на количественное содержание и стерильность |
Таблица составлена авторами по материалам Резолюции Рабочей группы фармацевтов Германии (Arbeitsgemeinschaft der Pharmazieräte Deutschlands) от 16.10.2013
Примечание. ЛФ — лекарственная форма; АФС — фармацевтическая субстанция; ЛС — лекарственное средство.
Резолюция APD 2013 устанавливает следующий возможный подход АО при внутриаптечном контроле ВАЗ:
Несколько иную классификацию представляет Формуляр DAC/NRF, который определяет три уровня риска: низкий, средний и высокий. Для определения соответствующего уровня риска ВАЗ используются критерии Резолюции ResAP, где степень риска оценивается за счет произведения значений коэффициентов (табл. 5. «Форма для оценки риска внутриаптечной заготовки согласно Формуляру DAC/NRF». Опубликована на сайте журнала)28.
Согласно Формуляру DAC/NRF для ВАЗ с низким уровнем риска используются простые методы физического и химического анализа по определению простых аналитических характеристик, со средним уровнем — простые методы физического и химического анализа, а также методы полного химического контроля (качественный и количественный анализ для действующих веществ у некоторых ЛФ), с высоким — ко всему перечисленному дополнительно осуществляется количественное определение АФС и ВВ.
При разработке методов испытаний и заданных значений заведующий аптекой должен использовать следующие источники информации: Фармакопею США, Британскую или Швейцарскую фармакопеи, монографии «стандартных разрешений», действующую Немецкую фармакопею, Формуляр DAC/NRF, научные публикации в рецензируемых и специализированных изданиях.
В соответствии с Формуляром DAC/NRF количество отбираемых упаковок ВАЗ (N) рассчитывается по формуле (1):
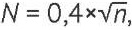 , (1)
, (1)
где n — количество упаковок одной серии ВАЗ.
Приведенная формула аналогична ОФС.1.1.0004 «Отбор проб» Государственной фармакопеи Российской Федерации XV издания, однако для малых серий Формуляром DAC/NRF предусмотрены соответствующие округления, где N составляет: до 1,49 — 1 упаковка; от 1,50 до 2,49 — 2 упаковки; от 2,50 до 3,49 — 3 упаковки; >3,50 — 4 упаковки.
В случае использования стандартных рецептур применяются указанные в Формуляре DAC/NRF методики контроля качества.
Несмотря на то что при изготовлении ВАЗ увеличиваются требования к контролю качества, как правило, себестоимость изготовления ВАЗ в пересчете на одну упаковку, по сравнению с РЭЛП, ниже. Пример затрат на изготовление ЛС представлен в таблице 6 «Сравнение стоимости изготовления внутриаптечной заготовки и составных лекарственных средств на примере изготовления 20 упаковок гидрофильного крема бетаметазона валерат 0,1% — 50,0 (Формуляр DAC/NRF, рецептура NRF 11.37), в ценах 2019 г.» (опубликована на сайте журнала)29.
Блок-схема изготовления ВАЗ представлена на рисунке 2 «Блок-схема изготовления внутриаптечной заготовки» (опубликован на сайте журнала на английском языке30).
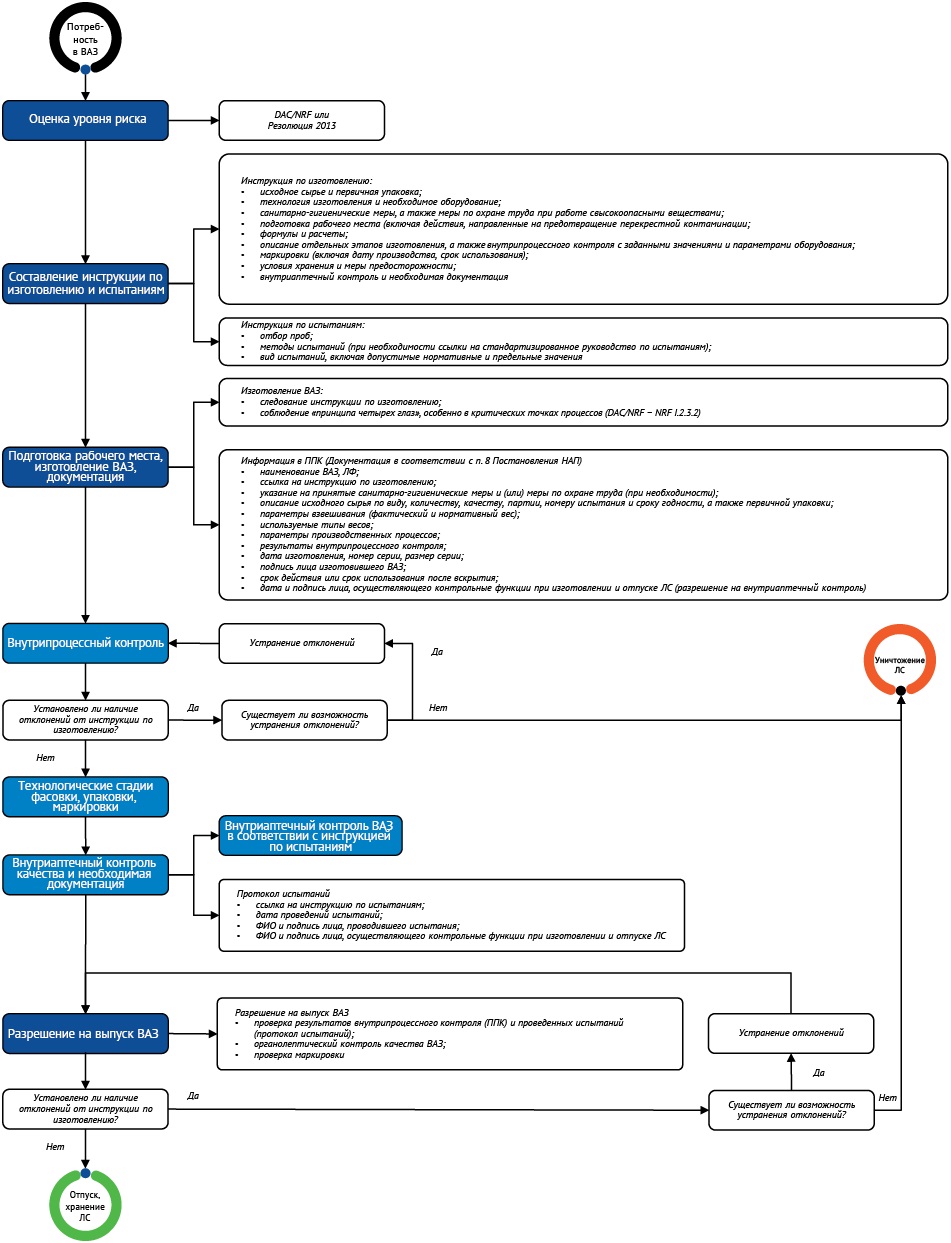
Рисунок подготовлен авторами / The figure is prepared by the authors
Рис. 2. Блок-схема изготовления внутриаптечной заготовки (ВАЗ). DAC/NRF — национальный формуляр (Deutscher Arzneimittel-Codex / Neues Rezeptur-Formularium); ППК — паспорт письменного контроля; НАП — постановление Минздрава Германии «О работе аптечных организаций» от 09.02.1987; ЛС — лекарственное средство
Fig. 2. Flowchart for compounding stock preparations30
Согласно п. 11 Постановления НАП используемые при изготовлении АФС должны быть надлежащего качества, которое может быть проверено в производственной аптеке либо за ее пределами (п. 3 Постановления НАП). При использовании АФС, качество которых подтверждено сертификатом испытаний сторонней организации, при приемочном контроле осуществляется определение подлинности. Испытаниям также подвергаются все изготовленные в АО тритурации, концентраты, полуфабрикаты, а результаты испытаний оформляются протоколом испытаний (п. 8 Постановления НАП).
В соответствии с п. 11а Постановления НАП АО может передать на аутсорсинг производителям ЛС или другой производственной аптеке деятельность по изготовлению (восстановлению) следующих ЛС: цитостатических ЛС; ЛС для парентерального питания; ЛС, применяемых в других, обоснованных с медицинской точки зрения, случаях особой необходимости; расфасованные ГЛФ; ЛС, которые применяются для лечения угрожающего инфекционного заболевания, распространение которого требует немедленной поставки конкретных ЛС, значительно превышающих обычный уровень потребности. Для этого должен быть заключен письменный договор оказания аутсорсинговых услуг. Руководитель аптеки не может заказывать изготовление ЛС без наличия рецепта врача.
ГЛФ, реализуемые в АО, подлежат выборочному органолептическому контролю качества (п. 12 Постановления НАП). При наличии сомнений в качестве ГЛФ необходимо провести их полный химический контроль.
Протокол испытаний ГЛФ должен содержать название производителя; наименование и ЛФ; номер серии и дату производства; дату и результаты проверки; ФИО и подпись фармацевта, осуществившего контроль качества.
Требования к маркировке РЭЛП установлены п. 14 Постановления НАП.
Особо следует отметить, что допускается изготовление парентеральных ЛФ из ГЛФ, при этом в значительной части случаев их изготавливают только из ГЛФ, что также предусмотрено тарифами на разведение (восстановление) ЛС. Если вместо АФС использовалась ГЛФ, достаточно указать только ее состав. В случае если ЛС изготовлен в парентеральной ЛФ, указывается состав ГЛФ, торговое наименование ГЛФ и производителя, номер серии. ВАЗ, предназначенная для отпуска, маркируется как ГЛФ (ст. 10 Закона Германии).
Согласно п. 17 Постановления НАП автоматизированные пункты выдачи ЛС (вендинговые аппараты) разрешено размещать исключительно в АО. Доставка ЛС осуществляется курьерами производственной аптеки без дополнительной лицензии. При осуществлении курьерской доставки ЛС заведующий аптекой должен обеспечить надлежащее соблюдение условий хранения ЛС, кроме того, должна существовать система отслеживания отправлений. Доставка должна осуществляться фармацевтическими работниками, если до осуществления доставки в АО не был представлен рецепт на рецептурные ЛС или не была проведена фармацевтическая консультация.
АО имеют право изготавливать ЛС и отпускать ГЛФ для ветеринарного применения. Согласно п. 19 Постановления НАП для осуществления такой деятельности необходимо вести учет реализованных рецептурных ЛС для ветеринарного применения.
В соответствии с п. 24 Постановления НАП при отсутствии АО в отдельном районе Германии может быть открыт пункт приема рецептов для изготовления ЛС. Разрешение на деятельность пункта приема является временным и действует в течение трех лет. Рецепты собирают в контейнер, который опечатывается при доставке в АО. На маркировке контейнера указывается название и адрес аптеки, время получения, а также способ получения ЛС пациентом (самовывоз или доставка).
По смыслу п. 26 Постановления НАП к больничным аптекам относят структурные подразделения МО, в обязанности которых входит обеспечение надлежащего снабжения одной или нескольких больниц (межбольничные аптеки) ЛС и медицинскими изделиями. Требования к помещениям больничной аптеки установлены в п. 29 Постановления НАП: площадь производственного помещения не менее 200 м², две ассистентских комнаты с вытяжными шкафами, служебное и подсобное помещение, помещения для хранения исходного сырья и ЛС. Согласно п. 33 Постановления НАП все больничные аптеки должны работать круглосуточно.
При осуществлении деятельности по индивидуальной фасовке ЛС в состав СМК должны входить: перечни ГЛФ, разрешенных к фасовке; перечни ГЛФ, запрещенные к фасовке в одном контейнере; инструкция по хранению и маркировке ГЛФ, изъятых из первичной упаковки; требования к калибровке, квалификации, техническому обслуживанию и очистке оборудования для фасовки; описание используемых упаковочных материалов; инструкция по фасовке; инструкция по санитарному режиму; гигиенические требования к персоналу и средствам индивидуальной защиты. Помещение для индивидуальной фасовки должно быть достаточного размера для проведения отдельных операций. Доступ к помещению и внесение в него исходных материалов должно осуществляться через промежуточное пространство (шлюз), чтобы обеспечить соответствующее качество воздуха в помещении.
Согласно п. 35 Постановления НАП при изготовлении парентеральных ЛФ СМК производственных аптек должны включать в себя как минимум следующие сведения: об используемом исходном сырье, а также о первичных упаковочных материалах и результатах проверки их качества; о технических и организационных мерах по предотвращению микробной и перекрестной контаминаций; о калибровке, квалификации, обслуживании и очистке оборудования и помещений; о валидации процессов, методов, методик и систем, влияющих на качество ЛС, а также ревалидации; о критически важном оборудовании или устройствах; инструкции по изготовлению, испытаниям, а также о возможной транспортировке ЭЛП; о проведении комплекса санитарно-гигиенических мероприятий.
Изготовление парентеральных ЛФ должно производиться в отдельном помещении, которое не может быть использовано для осуществления других видов деятельности, за исключением изготовления других стерильных ЛФ в соответствии с Фармакопеей. Доступ в асептический блок, а также подача исходного сырья, упаковочных материалов должны осуществляться через воздушный шлюз. Площадь помещения должна обеспечивать выполнение отдельных операций на специальных рабочих местах. Во время изготовления в помещении могут находиться только сотрудники в защитной одежде, предотвращающей контаминацию ЭЛП. Уровень чистоты помещения определяется в зависимости от порядка изготовления и наличия процесса стерилизации ЛС в соответствии с Правилами надлежащей производственной практики Европейского союза31, а вентиляция должна быть обеспечена фильтрами достаточной эффективности. Если ЭЛП не подвергаются стерилизации в конечной упаковке и не изготавливаются с использованием изоляторных технологий, во время приготовления и розлива в локальной зоне для рабочих процессов устанавливается уровень чистоты воздуха по количеству микробов и частиц, соответствующий классу А, и внешняя среда с точки зрения количества частиц и микробов соответствует:
Для фасовки жидких ЛФ необходимо поддерживать чистоту воздуха класса C.
Условия в чистых помещениях должны проверяться путем определения количества твердых частиц и микробов в воздухе, на критических поверхностях и проверки персонала во время изготовления в открытых системах.
Требования по микробиологической чистоте помещений при изготовлении нестерильных ЛС не установлены законодательством Германии.
Фармаконадзором ЛС занимается Комиссия фармацевтов Германии (нем. Arzneimittelkommission der Deutschen Apotheker), представляющая собой независимое национальное учреждение. Комиссия является частью Федерального союза немецких ассоциаций фармацевтов [1].
Для полноценной проверки качества ЛС, изготавливаемых в АО, общественные аптеки могут передать изготовленные ЛС в «Центральную лабораторию немецких фармацевтов» (нем. Zentrallaboratorium DeutscherApotheker e.V.), которая проводит полный контроль качества изготовленных ЛС, ежегодно исследует стабильность и, при необходимости, вносит изменения в сроки годности рецептур Формуляра DAC/NRF.
Проведенное исследование позволяет выделить следующие значимые особенности организации деятельности по изготовлению ЛС на территории Германии.
В 2023 г. в Российской Федерации завершилась работа по формированию подзаконного законодательства в отношении принятого Федерального закона № 502-ФЗ «О внесении изменений в статью 56 Федерального закона «Об обращении лекарственных средств». Тем не менее разработанные документы и ряд их положений, в том числе концептуальных, сохранили свою преемственность с организационно-фармацевтическими подходами, отраженными в нормативных правовых актах, созданных в Советском Союзе до 1968 г., что в том числе представлено в ранее проведенных исследованиях [2].
25 июня 2024 г. на очередном заседании рабочей группы по производственным аптекам при Комитете Государственной Думы по охране здоровья, а также в рамках рабочего совещания с участием Минздрава России и Фонда «Круг добра», состоявшегося 8 июля 2024 г., была окончательно подтверждена и определена роль производственных аптек для отечественной системы здравоохранения. В частности, сформулирована одна из ключевых задач аптечной производственной инфраструктуры как критически значимого элемента лекарственного обеспечения лиц с тяжелыми жизнеугрожающими и хроническими заболеваниями, в том числе редкими (орфанными) заболеваниями.
В заключение следует отметить, что в случае установленных соответствующих целей, задач, мероприятий и целевых показателей в новых национальных проектах, государственных программах развития здравоохранения, программе государственных гарантий — приоритета системы здравоохранения по развитию в своей структуре современной, высокотехнологичной инфраструктуры по изготовлению ЛП, перехода к принципам персонализированной медицины, внедрения фармакоэкономических моделей, основанных на принципах курсовых назначений при оказании медицинской помощи и лекарственном обеспечении населения, российскому медицинскому, фармацевтическому сообществу и отраслевому законодательству предстоит путь от принятия концепции регулирования до внедрения самых передовых решений в клиническую практику, где ключевым шагом будут выступать поправки в ряд федеральных законов и, прежде всего, в ст. 56 ФЗ-61, в том числе в качестве установленного перехода от Правил изготовления и отпуска ЛП на Правила надлежащей практики изготовления и отпуска ЛП.
Дополнительная информация. На сайте журнала «Регуляторные исследования и экспертиза лекарственных средств» размещены таблицы 1–6, рис. 1 и 2.
https://doi.org/10.30895/1991-2919-2024-590-table1
https://doi.org/10.30895/1991-2919-2024-590-table2
https://doi.org/10.30895/1991-2919-2024-590-table3
https://doi.org/10.30895/1991-2919-2024-590-table4
https://doi.org/10.30895/1991-2919-2024-590-table5
https://doi.org/10.30895/1991-2919-2024-590-table6
https://doi.org/10.30895/1991-2919-2024-590-fig1
https://doi.org/10.30895/1991-2919-2024-590-fig2
Additional information. Tables 1–6, and Figures 1, 2 are published on the website of Regulatory Research and Medicine Evaluation.
https://doi.org/10.30895/1991-2919-2024-590-table1
https://doi.org/10.30895/1991-2919-2024-590-table2
https://doi.org/10.30895/1991-2919-2024-590-table3
https://doi.org/10.30895/1991-2919-2024-590-table4
https://doi.org/10.30895/1991-2919-2024-590-table5
https://doi.org/10.30895/1991-2919-2024-590-table6
https://doi.org/10.30895/1991-2919-2024-590-fig1
https://doi.org/10.30895/1991-2919-2024-590-fig2
Вклад авторов. Все авторы подтверждают соответствие своего авторства критериям ICMJE. Все авторы внесли существенный вклад в разработку концепции, проведение исследования и подготовку статьи, прочли и одобрили финальную версию статьи перед публикацией.
Authors’ contributions. All the authors confirm that they meet the ICMJE criteria for authorship. All the authors have made substantial contributions to the conception of the work, the conduct of the research, and the preparation of the article. All the authors have read and approved the final version prior to its publication.
1. https://www.gesetze-im-internet.de
2. https://dejure.org/
3. В России Национальной ассоциацией управленцев сферы здравоохранения разработан законопроект об индивидуальном правовом статусе практикующего врача, согласно которому такой статус смогут получать специалисты с высшим или средним специальным медицинским образованием, стажем работы по специальности не менее пяти лет. Ведение соответствующего реестра медицинских работников будет осуществлять Федеральная медицинская палата (Калиновский И. В Госдуму направят законопроект об индивидуальном правовом статусе врача. Медвестник. 10.04.2024.
https://medvestnik.ru/content/news/V-Gosdumu-napravyat-zakonoproekt-ob-individualnom-pravovom-statuse-vracha.html)
4. Bundesärzteordnung. https://www.gesetze-im-internet.de/b_o/index.html
5. Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz — AMG). https://www.gesetze-im-internet.de/amg_1976/BJNR024480976.html
6. Leitsätze zum Urteil des Ersten Senats vom 16.02.2000. 1 BvR 420/97. https://www.bundesverfassungsgericht.de/SharedDocs/Entscheidungen/DE/2000/02/rs20000216_1bvr042097.html
7. Grundgesetz für die Bundesrepublik Deutschland. https://www.gesetze-im-internet.de/gg/BJNR000010949.html
8. https://www.bundesrat.de/DE/plenum/themen/foekoI/bundesstaatskommission/unterlagen/AU-002.pdf?__blob=publicationFile&v=1
9. Gesetz über das Apothekenwesen. https://www.gesetze-im-internet.de/apog/__2.html
10. Verordnung über den Betrieb von Apotheken (Apothekenbetriebsordnung — ApBetrO). https://www.gesetze-im-internet.de/apobetro_1987/ApBetrO.pdf
11. Anzeige gemäß § 67 Absatz 1 und 2 Arzneimittelgesetz (AMG) für die erlaubnisfreie Herstellung von Arzneimitteln durch ärztliche, zahnärztliche sowie andere zur Ausübung der Heilkunde bei Menschen befugte Personen gemäß § 13 Absatz 2b AMG und die erlaubnisfreie Herstellung von Gewebe und Gewebezubereitungen durch ärztliche Personen gemäß § 20d AMG. https://www.berlin.de/lageso/gesundheit/pharmaziewesen-und-medizinprodukte/arzneimittelwesen/vorlage_anzeige_-67.docx?ts=1685105902
12. OLG Hamburg, Urteil vom 18.12.2015 — 3 U 43/14. https://openjur.de/u/934358.html
13. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A02001L0083-20220101
14. InfoCuria Case-law. 16.07.2015 — C-544/13 — Abcur. https://curia.europa.eu/juris/liste.jsf?&num=C-544/13
15. LG Hamburg, Urteil vom 04.02.2021 — 312 O 112/20. https://openjur.de/u/2333565.html
16. Vierte Verordnung zur Änderung der Apothekenbetriebsordnung. http://www.bundesrat.de/drs.html?id=61-12
17. ГОСТ Р ИСО 9001-2015. Системы менеджмента качества.
18. DAC/NRF. https://dacnrf.pharmazeutische-zeitung.de/
19. https://doi.org/10.30895/1991-2919-2024-590-table1
20. Здесь и далее по тексту «срок годности» — собирательный термин, включающий срок действия и срок использования после вскрытия, поскольку аптечная организация должна указать эти данные в зависимости от вида экстемпорального лекарственного препарата.
21. https://doi.org/10.30895/1991-2919-2024-590-table2
22. Приказ Министерства здравоохранения Российской Федерации от 22.05.2023 № 249н «Об утверждении правил изготовления и отпуска лекарственных препаратов для медицинского применения аптечными организациями, имеющими лицензию на фармацевтическую деятельность».
23. Англоязычная версия рисунка 1 размещена на сайте журнала (English translation of Figure 1 is available at the website of the journal). https://doi.org/10.30895/1991-2919-2024-590-fig1
24. https://doi.org/10.30895/1991-2919-2024-590-table3
25. Resolution 2013. https://www.pharmazierat.de/logicio/pmws/indexDOM.php?client_id=pharmazierat&page_id=resolutionen2013&lang_iso639=de
26. Англоязычная версия таблицы 4 размещена на сайте журнала (English translation of Table 1 is available at the website of the journal). https://doi.org/10.30895/1991-2919-2024-590-table4
27. Verordnung über den Betrieb von Apotheken (Apothekenbetriebsordnung — ApBetrO). https://www.gesetze-im-internet.de/apobetro_1987/ApBetrO.pdf
28. Таблица 5 размещена на сайте журнала (Table 5 is available at the website of the journal). https://doi.org/10.30895/1991-2919-2024-590-table5
29. Таблица 6 размещена на сайте журнала (Table 6 is available at the website of the journal). https://doi.org/10.30895/1991-2919-2024-590-table6
30. Англоязычная версия рисунка 2 размещена на сайте журнала (English translation of Figure 2 is available at the website of the journal). https://doi.org/10.30895/1991-2919-2024-590-fig2
31. EudraLex — Volume 4 — Good Manufacturing Practice (GMP) guidelines. https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en
1. Эрдни-Гаряев СЭ, Мамедов ДД, Юрочкин ДС, Зеликова ДД, Голант ЗМ, Фисенко ВС, Наркевич ИА. Нормативное правовое регулирование изготовления лекарственных препаратов аптечными организациями на немецком фармацевтическом рынке. Часть 1. Основные положения законодательства (обзор). Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):91–109. https://doi.org/10.30895/1991-2919-2024-14-1-91-109
2. Наркевич ИА, Фисенко ВС, Голант ЗМ, Юрочкин ДС, Мамедов ДД, Эрдни-Гаряев СЭ, Лешкевич АА. Основы формирования единой гармонизированной системы нормативного правового регулирования в области обращения лекарственных препаратов, изготавливаемых аптечными организациями. СПб: Медиапапир; 2023. EDN: PZEVDF
3. Фаррахов АЗ, Соломатина ТВ, Мамедов ДД, Юрочкин ДС, Голант ЗМ, Наркевич ИА. Основы формирования стратегии развития сегмента изготовления и отпуска лекарственных препаратов в Российской Федерации. Вестник Росздравнадзора. 2023;(6):6–17. EDN: NAHOAC
4. Bouwman-Boer Y, Fenton-May V, Le Brun P, eds. Practical pharmaceutics. An international guideline for the preparation, care and use of medicinal products. Cham, Switzerland: Springer; 2015. https://doi.org/10.1007/978-3-319-15814-3
Эрдни-Гаряев Сергей Эдуардович
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022
Мамедов Деви Девивич
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022
Юрочкин Дмитрий Сергеевич
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022
Зеликова Дарья Дмитриевна
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022
Голант Захар Михайлович, канд. экон. наук
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022
Фисенко Виктор Сергеевич, канд. фарм. наук
Рахмановский пер., д. 3, ГСП 4, Москва, 127994
Наркевич Игорь Анатольевич, д-р фарм. наук, профессор
ул. Профессора Попова, 14, лит. А, Санкт-Петербург, 197022

|
1. Таблица 1. Пример установления срока использования исходного сырья в аптечной организации Германии | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(909KB)
|
Метаданные ▾ | |
|
|
2. Таблица 2. Максимальные сроки годности некоторых лекарственных форм экстемпоральных лекарственных препаратов в Германии | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(933KB)
|
Метаданные ▾ | |
|
|
3. Таблица 3. Критерии принятия решения при оценке риска | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(926KB)
|
Метаданные ▾ | |
|
|
4. Table 4. Risk categories and internal quality control examples | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
5. Таблица 5. Форма для оценки риска внутриаптечной заготовки согласно Формуляру DAC/NRF | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(815KB)
|
Метаданные ▾ | |
|
|
6. Таблица 6. Сравнение стоимости изготовления внутриаптечной заготовки и составных лекарственных средств на примере изготовления 20 упаковок гидрофильного крема бетаметазона валерат 0,1% — 50,0 (Формуляр DAC/NRF, рецептура NRF 11.37.), в ценах 2019 г. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
7. Fig. 1. Flowchart for compounding extemporaneous formulations | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
8. Fig. 2. Flowchart for compounding stock preparations | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
Эрдни-Гаряев С.Э., Мамедов Д.Д., Юрочкин Д.С., Зеликова Д.Д., Голант З.М., Фисенко В.С., Наркевич И.А. Нормативное правовое регулирование изготовления лекарственных препаратов аптечными организациями на немецком фармацевтическом рынке. Часть 2. Особенности организации деятельности (обзор). Регуляторные исследования и экспертиза лекарственных средств. 2025;15(1):63-81. https://doi.org/10.30895/1991-2919-2024-590
Erdni-Garyaev S.E., Mamedov D.D., Yurochkin D.S., Zelikova D.D., Golant Z.M., Fisenko V.S., Narkevich I.A. Pharmacy Compounding Regulation in the German Pharmaceutical Market. Part 2. Organisational Features (Review). Regulatory Research and Medicine Evaluation. 2025;15(1):63-81. (In Russ.) https://doi.org/10.30895/1991-2919-2024-590

Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных




























