




Содержание
Перейти к:
https://doi.org/10.30895/1991-2919-2023-549
Перейти к:
АКТУАЛЬНОСТЬ. Важными задачами при создании лекарственных средств являются формирование в области фармацевтической разработки системы менеджмента качества (СМК) и разработка инструментов оценки ее эффективности.
ЦЕЛЬ. Предложить модель системы менеджмента качества в области фармацевтической разработки биотехнологических препаратов, а также разработать механизм оценки эффективности функционирования данной системы в различных подразделениях научно-исследовательского центра.
МАТЕРИАЛЫ И МЕТОДЫ. В работе представлены данные о результатах внутренних аудитов научно-исследовательского центра, результаты работы с несоответствиями и данные о документообороте. Значения параметров для расчета индекса качества регистрировали в валидированных Excel-файлах. Для анализа и визуализации данных использовали программный продукт Microsoft Power BI (Business Intelligence), предназначенный для бизнес-анализа данных. Основными используемыми компонентами платформы Power BI являлись Power BI Desktop и Power BI Service.
РЕЗУЛЬТАТЫ. Внедрена СМК и разработан механизм, позволяющий проводить оценку эффективности работы СМК в подразделениях научно-исследовательского центра вне зависимости от индивидуальных требований к каждому подразделению.
ВЫВОДЫ. Предложены модель организации СМК в области фармацевтической разработки биотехнологических препаратов и инструмент (индекс качества) для сбора оцифрованных данных, проведения унифицированного мониторинга и отслеживания динамики изменения состояния СМК вне зависимости от индивидуальных требований к каждому подразделению. Отмечено, что индекс качества может быть применен не только в отношении подразделений разработки лекарственных препаратов, но и в отношении других научных подразделений ввиду его универсальности, простоты и возможности модификации за счет введения дополнительных индивидуальных показателей качества.
Гиба И.С., Салиева К.Р., Батуева А.А., Григорьева И.В., Драй Р.В. Внедрение и оценка системы менеджмента качества в области фармацевтической разработки биотехнологических лекарственных препаратов. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):62-71. https://doi.org/10.30895/1991-2919-2023-549
Giba I.S., Salieva K.R., Batueva A.A., Grigorieva I.V., Drai R.V. Quality Management in Pharmaceutical Development of Biotechnology-Derived Medicinal Products: Implementation and Evaluation. Bulletin of the Scientific Centre for Expert Evaluation of Medicinal Products. Regulatory Research and Medicine Evaluation. 2024;14(1):62-71. (In Russ.) https://doi.org/10.30895/1991-2919-2023-549
В связи с возрастающей многополярностью мира, необходимостью укрепления национального суверенитета, а также учитывая возможность возникновения и быстрого распространения эпидемий, остро стоит вопрос об ускорении фармацевтической разработки и быстром выводе лекарственных препаратов на рынок. Развитие системы менеджмента качества (СМК), позволяющей обеспечить целостность данных в процессе разработки и производства лекарственного препарата (ЛП), а также систематизировать работу сотрудников в области применения новых методов исследований при разработке ЛП, является актуальной задачей.
Система менеджмента качества на фармацевтическом производстве должна соответствовать требованиям государственных1 и международных2 организаций, рекомендациям Всемирной организации здравоохранения (ВОЗ)3, Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения (International Council for Harmonisation of Technical Requirements for Pharmaceutical for Human Use, ICH)4 и Конвенции по фармацевтическим инспекциям (Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme, PIC/S)5. Инструменты и методы приведения СМК в соответствие с этими требованиями описаны в документах Международной ассоциации фармацевтического инжиниринга (International Society for Pharmaceutical Engineering, ISPE), Ассоциации производителей парентеральных лекарственных средств (Parenteral Drug Association, PDA), Американского общества по испытанию материалов (American Society for Testing and Materials, ASTM) и др. В периодических изданиях публикуются как рекомендации представителей регуляторных органов по развитию СМК в области производства лекарственных средств и медицинских изделий [1–4], так и результаты научных исследований по данной теме [5–7]. В последние годы произошла смена парадигмы фармацевтической разработки: вместо подхода, ориентированного на испытания конечного продукта (Quality by Test, QbT), используют подход, направленный на обеспечение качества лекарственных средств путем управления рисками на всех этапах производства (Quality by Design, QbD) [8–12], рекомендованный ICH в 2009 г.6
Однако принципы и требования к организации СМК в области фармацевтической разработки описаны достаточно поверхностно7, а конкретные механизмы внедрения СМК и оценки ее надлежащего функционирования не описаны вовсе [13, 14].
Важность развития этого направления неоднократно подчеркивалась ВОЗ, и в момент написания данной статьи рекомендации по надлежащей практике фармацевтической разработки находились на этапе формирования8. Учитывая, что научные данные, полученные в ходе фармацевтической разработки, входят в Общий технический документ (Common Technical Document, CTD) модуля 3 «Качество», переоценить их значение для выведения лекарственного препарата на рынок невозможно.
Сложность создания СМК в области фармацевтической разработки и оценки ее эффективности, по мнению авторов, заключается в невозможности унифицировать требования к результатам работы различных производственных подразделений, аналитических лабораторий и научно-исследовательских центров, деятельность которых регулируется различными нормативными документами. Так, в разработке биотехнологических препаратов принимают участие не только подразделения, которые занимаются ведением штаммов продуцентов, определением критических параметров процессов (Critical Process Parameters, CPPs) ферментации, выделения, очистки, получения готовых лекарственных форм, определением критических показателей качества (Critical Quality Attributes, CQAs), но и подразделения, в сферу деятельности которых входит анализ образцов сырья и полупродуктов согласно требованиям ISO:170259, а также разработка и валидация аналитических методик контроля качества, включая трансфер методик на производственные площадки. Также в разработке ЛП участвуют подразделения, проводящие доклинические исследования в соответствии с принципами надлежащей лабораторной практики (Good Laboratory Practice, GLP)10.
В настоящее время модели формирования СМК и инструменты ее оценки в области фармацевтической разработки на государственном уровне отсутствуют, несмотря на отдельные попытки создать международные организации, уполномоченные решать подобные вопросы11.
Цель работы — предложить модель системы менеджмента качества в области фармацевтической разработки биотехнологических препаратов, а также разработать универсальный механизм оценки эффективности функционирования данной системы в различных подразделениях научно-исследовательского центра.
Для достижения поставленной цели были определены следующие задачи.
Внедрение СМК и универсальной метрики для
оценки и анализа было реализовано в подразделениях R&D центра ООО «ГЕРОФАРМ», занимающегося разработкой биотехнологических препаратов. В состав центра входили следующие подразделения (лаборатории):
1) лаборатория культивирования (Upstream Process, USP);
2) лаборатория выделения и очистки (Downstream Process, DSP);
3) лаборатория разработки готовых форм (Finished Dosage Form, FDF);
4) физико-химическая лаборатория (Chemical Physics Laboratory, CPL);
5) микробиологическая лаборатория (Microbiological Laboratory, MBL).
Задача лаборатории USP — разработка дизайна и получение генетических конструкций, создание штаммов-продуцентов, фармацевтическая разработка технологий культивирования. Задача лаборатории DSP — разработка процессов выделения и очистки, таких как хроматография, вирусная инактивация, вирусная фильтрация, диафильтрация и др. Задача лаборатории FDF — разработка состава готовой лекарственной формы, технологии производства, спецификаций, подтверждение сопоставимости препарата до и после изменения производственного процесса12 и стабильности готового продукта при хранении. СМК в лабораториях USP, DSP и FDF была организована согласно ICH Q8 (R2)13 и ICH Q1114.
Задачи лаборатории CPL — разработка и валидация аналитических методик, используемых для контроля качества полупродуктов и готового продукта физико-химическими методами (спектральные, оптические, хроматографические, электрохимические), анализ образцов, полученных от USP, DSP и FDF в ходе фармацевтической разработки, а также проведение исследований сопоставимости и стабильности. СМК в подразделении CPL была организована согласно ГОСТ ISO/IEC 17025-201915 и Приказу № 70716.
Задачи лаборатории MBL — разработка и валидация микробиологических методик для контроля качества полупродуктов и готового продукта, анализ образцов, полученных от USP, DSP и FDF в ходе фармацевтической разработки и проведение доклинических исследований. СМК в подразделении MBL была организована согласно принципам GLP17.
В каждой из лабораторий предварительно были разработаны и внедрены стандартные операционные процедуры (Standard Operation Procedures, SOPs), стандартные заполняемые формы и шаблоны, в совокупности описывающие весь процесс фармацевтической разработки.
Анализ СМК включал следующие направления работы:
1) внутренние аудиты (инспекции);
2) система работы с несоответствиями;
3) документооборот.
Внутренние аудиты (инспекции). Для каждого подразделения был составлен годовой план внутренних аудитов (инспекций). В соответствии со спецификой СМК аудиты (инспекции) подразделялись на несколько типов.
Для подразделений USP, DSP и FDF проводились:
Для CPL:
Для MBL:
Каждому аудиту (инспекции) присваивали индивидуальный номер кодирования: InA_XX, где XX — порядковый номер аудита (инспекции). Аудиты (инспекции) проводились службой обеспечения качества — отдельным независимым подразделением, не вовлеченным в разработку биотехнологических препаратов.
Работа с несоответствиями. Несоответствия, выявленные в ходе внутреннего аудита (инспекции), классифицировали по категориям: критические, значительные и незначительные. Для подразделений USP, DSP, FDF и CPL классификация была проведена следующим образом:
Для подразделения MBL несоответствия требованиям GLP классифицируются следующим образом:
По результатам аудита для устранения выявленных несоответствий сотрудники подразделений составляли план корректирующих и предупреждающих действий (Corrective and Preventive Action, CAPA) с указанием сроков выполнения. Руководитель подразделения утверждал план CAPA и передавал информацию в службу обеспечения качества, сотрудники которой проверяли эффективность реализации плана CAPA после устранения всех несоответствий при проведении очередного планового аудита (инспекции).
Документооборот. Для каждого подразделения был составлен годовой план разработки и пересмотра внутренних документов, включая SOPs, определены планируемые сроки введения и ответственные за разработку.
Индекс качества. Для универсальной оценки эффективности СМК подразделений, осуществляющих разработку биотехнологических лекарственных препаратов, было предложено использовать индекс качества (q), позволяющий учесть результаты внутренних аудитов (инспекций), работу над устранением несоответствий по планам CAPA и отношение SOPs, не введенных и введенных с нарушением сроков, к общему числу запланированных SOPs. Расчет q выполняли по формуле (1):
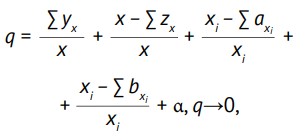 (1)
(1)
где x — число аудитов (инспекций) за анализируемый период; ∑yx — суммарное превышение норм несоответствий (отсутствие превышений y = 0, более пяти незначительных несоответствий y = 1, более двух значительных несоответствий y = 2, наличие критических несоответствий y = 3); ∑zx — сумма коэффициентов отклонений от норм по утверждению планов CAPA (не более 10 рабочих дней согласно внутренним процедурам R&D центра: при отсутствии отклонений от норм по утверждению планов CAPA или при отсутствии планов CAPA, если в результате аудита несоответствия не зафиксированы, z = 1, при наличии отклонений z = 0); xi — число планов CAPA, по которым не были выполнены все запланированные корректирующие действия; ∑axi — сумма коэффициентов отклонения от утвержденных сроков выполнения корректирующих действий по планам CAPA (отсутствие отклонений α = 1, нарушение сроков выполнения корректирующих действий α = 0); ∑bxi – сумма коэффициентов отклонения от сроков выполнения корректирующих действий по планам CAPA на конец анализируемого периода (корректирующие действия выполнены на конец анализируемого периода b = 1, корректирующие действия не выполнены на конец анализируемого периода b = 0); α — отношение SOPs, не введенных и введенных с нарушением сроков, к общему числу запланированных SOPs на анализируемый период.
Анализ данных для расчета индекса q проводился один раз в квартал. Формула (1) позволяет учесть результаты проведенных аудитов (инспекций) за квартал, превышение установленных норм количества несоответствий, сроки утверждения планов CAPA, эффективность работы подразделений по устранению несоответствий за анализируемый период, эффективность разработки и внедрения внутренних документов (SOPs).
Значение параметров для расчета q вносили в валидированные Excel-файлы. Для анализа и визуализации данных использовали программный продукт Microsoft Power BI (Business Intelligence), предназначенный для бизнес-анализа данных. Основным компонентом платформы является Power BI Desktop, включающий Power Query (система для подключения к источникам данных и предобработки данных), Power Pivot (система для создания модели связанных между собой данных из нескольких источников и расчетов) и Power View (система для создания визуализаций: графиков, таблиц, диаграмм и т.д.). Для просмотра разработанного и опубликованного графического интерфейса использовали Power BI Service с ежедневным обновлением данных в панели мониторинга при регулярном подключении к локальным источникам данных (Excel-файлам).
Количество проведенных в течение года аудитов (инспекций) представлено в таблице 1.
Вне зависимости от специфики деятельности к оценке работы подразделений были применены общие требования по допустимому количеству несоответствий, выявленных в ходе аудита (инспекции), а именно не более 0 критических, не более 2 значительных и не более 5 незначительных. Результаты аудитов (инспекций) и информация по устранению несоответствий представлены в таблице 2.
Количество критических, значительных и незначительных несоответствий, выявленных в результате аудита (инспекции) для подразделений, представлено на рисунке 1.
Сравнение значений величины α, указанной в формуле (1), между подразделениями позволяет установить эффективность работы отдела по планированию и разработке внутренней документации, в данном случае SOPs, вне зависимости от количества документов. Результат выполнения утвержденных планов за год указан в таблице 3.
Выводы о выполнении планов по разработке SOPs на основании данных в таблице 3 можно выразить в процентном отношении, например 100% выполнение плана зафиксировано в лаборатории CPL в 1 и 3 кварталах, 60% — в MBL в 3 квартале, а также невыполнение плана разработки документов в подразделении FDF на протяжении всего года. Результаты анализа эффективности СМК по индексу качества за год представлены в таблице 4.
Исходя из эмпирических значений, полученных по результатам аудитов за год, были установлены границы значений (рабочие зоны) индекса качества для отслеживания изменений состояния СМК подразделений с выделением «зеленой», «желтой» и «красной» зон:
Динамика изменения значений q за анализируемый период, в нашем случае это календарный год, а также границы значений представлены на рисунке 2.
В течение года после внедрения данной системы оценки эффективности СМК в выбранных подразделениях наблюдалась разная динамика изменения значения q (рис. 2). Сотрудники лаборатории USP, находившейся в первом квартале в «желтой» зоне при q = 2,75, в течение года предприняли ряд действий по снижению количества выявленных в ходе аудита несоответствий и выполнению плана CAPA для устранения выявленных несоответствий в установленные сроки, что позволило подразделению к концу четвертого квартала войти в «зеленую» зону эффективности со значением q = 0,70. Значения q для лаборатории DSP показали перманентное нахождение подразделения в «желтой» зоне, что свидетельствует о недостаточности или о неэффективности мер, предпринятых подразделением для улучшения значения индекса качества. В лаборатории FDF наблюдалась положительная тенденция по снижению значения q, которое в первом квартале составило 6,00 («красная» зона), а на конец четвертого квартала подразделение вышло на границу «желтой» и «зеленой» зон со значением q = 1,00. В следующем квартале при выполнении корректирующих мер можно ожидать выхода q FDF в «зеленую» зону. Значения q для CPL показали отрицательную динамику: в первом квартале эффективность работы подразделения находилась в «зеленой» зоне со значением q = 0, в период со второго по четвертый квартал значения q для CPL сместились в «желтую» зону, что говорит о необходимости проведения корректирующих мероприятий для улучшения работы СМК. Значения q для MBL демонстрировали положительную динамику: в первом квартале значение q находилось в «желтой» зоне и было равно 1, но уже во втором квартале q сместилось в «зеленую» зону и оставалось в ней три квартала. Такая тенденция говорит не только о хорошем состоянии СМК в MBL, но и о достаточности мер по поддержанию ее эффективности в течение значительного периода.
Единый подход к оценке состояния СМК позволил сравнить эффективность работы подразделений между собой вне зависимости от специфики их деятельности. Расчет индекса качества был использован как визуальная количественная метрика (рис. 2) надлежащего функционирования СМК в подразделениях.
Ежеквартальный общий анализ данных о проведенных внутренних аудитах, результатах устранения несоответствий согласно разработанным планам корректирующих и предупреждающих действий позволил руководителям подразделений и руководству R&D центра в нужный момент выявить проблемы и выделить ресурсы на их решение.
Таблица 1. Количество проведенных аудитов (инспекций)
Table 1. Number of audits (inspections) conducted
|
Подразделение |
Количество аудитов (инспекций) |
|
Лаборатория культивирования (USP) |
4 |
|
Лаборатория выделения и очистки (DSP) |
3 |
|
Лаборатория разработки готовых форм (FDF) |
2 |
|
Физико-химическая лаборатория (CPL) |
5 |
|
Микробиологическая лаборатория (MBL) |
12 |
|
Общее количество |
26 |
Таблица составлена авторами по собственным данным / The table is prepared by the authors using their own data
Таблица 2. Сводная таблица результатов аудитов (инспекций) и информации по устранению несоответствий
Table 2. Summary of audit (inspection) results showing the status of non-compliances
|
Подразделение |
Номер аудита |
Соблюдение сроков утверждения плана CAPA |
Действия по плану CAPA выполнены в срок |
План CAPA выполнен на конец квартала |
|
Лаборатория культивирования (USP) |
InA_03 |
Да |
Нет |
Да |
|
InA_09 |
Да |
Да |
– |
|
|
InA_16 |
Да |
Да |
– |
|
|
InA_25 |
Да |
Да |
– |
|
|
Лаборатория выделения и очистки (DSP) |
InA_06 |
Не применимо |
– |
– |
|
InA_13 |
Да |
Нет |
Да |
|
|
InA_15 |
Да |
–* |
–* |
|
|
Лаборатория разработки готовых форм (FDF) |
InA_05 |
Да |
Нет |
Нет |
|
InA_22 |
Да |
–* |
–* |
|
|
Физико-химическая лаборатория (CPL) |
InA_04 |
Да |
Нет |
Нет |
|
InA_10 |
Не применимо |
– |
– |
|
|
InA_18 |
Не применимо |
– |
– |
|
|
InA_21 |
Да |
Нет |
Нет |
|
|
InA_23 |
Да |
–* |
–* |
|
|
Микробиологическая лаборатория (MBL) |
InA_01 |
Не применимо |
– |
– |
|
InA_02 |
Да |
Нет |
Да |
|
|
InA_07 |
Да |
Да |
– |
|
|
InA_08 |
Не применимо |
– |
– |
|
|
InA_11 |
Да |
Да |
– |
|
|
InA_12 |
Не применимо |
– |
– |
|
|
InA_14 |
Не применимо |
– |
– |
|
|
InA_17 |
Не применимо |
– |
– |
|
|
InA_19 |
Да |
Да |
– |
|
|
InA_20 |
Не применимо |
– |
– |
|
|
InA_24 |
Не применимо |
– |
– |
|
|
InA_26 |
Да |
Да |
– |
Таблица составлена авторами по собственным данным / The table is prepared by the authors using their own data
Примечание. «Не применимо» — нет плана CAPA ввиду отсутствия выявленных в ходе аудита несоответствий; «–» — действия не проводились.
* Завершение действий по плану CAPA было запланировано на год, следующий за анализируемым календарным годом. Выполнение этих действий будет учтено в квартальном расчете q следующего года.
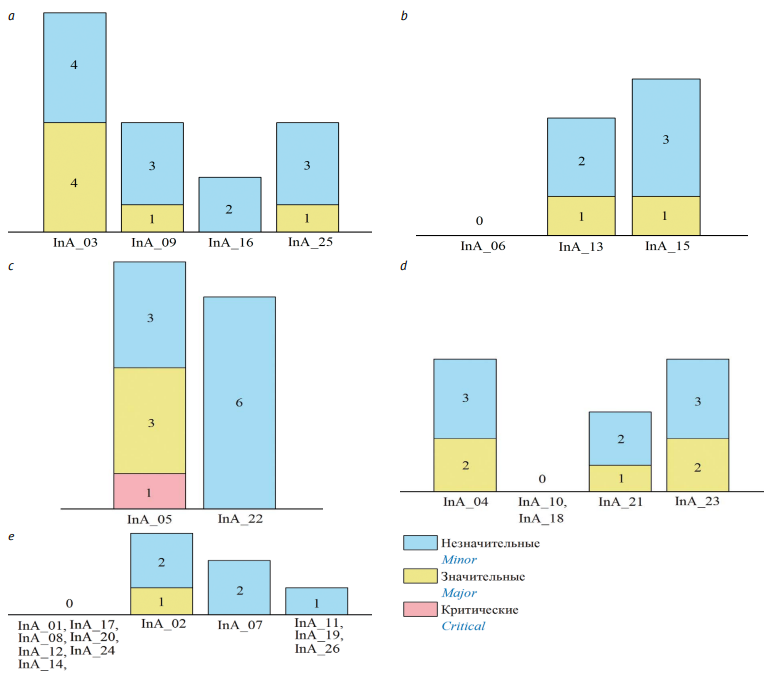
Рисунок подготовлен авторами по собственным данным / Figure is prepared by the authors using their own data
Рис. 1. Несоответствия, выявленные в результате аудитов (InA_... — номера аудитов): a — в лаборатории культивирования, b — в лаборатории выделения и очистки, c — в лаборатории разработки готовых форм, d — в физико-химической лаборатории, e — в микробиологической лаборатории
Fig. 1. Non-compliances identified during internal audits (InA_... show audit numbers) in the following divisions: a, Upstream Process; b, Downstream Process; c, Finished Dosage Form; d, Chemical Physics Laboratory; e, Microbiological Laboratory
Таблица 3. Результаты расчета величины α за год
Table 3. Results of calculating the α-value for 1 year
|
Подразделение |
Значение α |
|||
|
1 квартал |
2 квартал |
3 квартал |
4 квартал |
|
|
Лаборатория культивирования (USP) |
0,75 |
0,71 |
0,57 |
0,70 |
|
Лаборатория выделения и очистки (DSP) |
1,00 |
1,00 |
1,00 |
1,00 |
|
Лаборатория разработки готовых форм (FDF) |
1,00 |
1,00 |
1,00 |
1,00 |
|
Физико-химическая лаборатория (CPL) |
0,00 |
1,00 |
0,00 |
1,00 |
|
Микробиологическая лаборатория (MBL) |
1,00 |
0,50 |
0,40 |
0,64 |
Таблица составлена авторами по собственным данным / The table is prepared by the authors using their own data
Примечание. α — соотношение SOPs, не введенных и введенных с нарушением сроков, к общему числу запланированных SOPs на анализируемый период.
Таблица 4. Результаты расчета индекса качества (q) за год
Table 4. Results of calculating the quality index (q) for 1 year
|
Подразделение |
Универсальный индекс качества (q) |
|||
|
1 квартал |
2 квартал |
3 квартал |
4 квартал |
|
|
Лаборатория культивирования (USP) |
2,75 |
1,71 |
0,57 |
0,70 |
|
Лаборатория выделения и очистки (DSP) |
1,00 |
1,00 |
2,00 |
1,00 |
|
Лаборатория разработки готовых форм (FDF) |
6,00 |
3,00 |
3,00 |
1,00 |
|
Физико-химическая лаборатория (CPL) |
0,00 |
1,00 |
2,00 |
1,00 |
|
Микробиологическая лаборатория (MBL) |
1,00 |
0,50 |
0,40 |
0,64 |
Таблица составлена авторами по собственным данным / The table is prepared by the authors using their own data
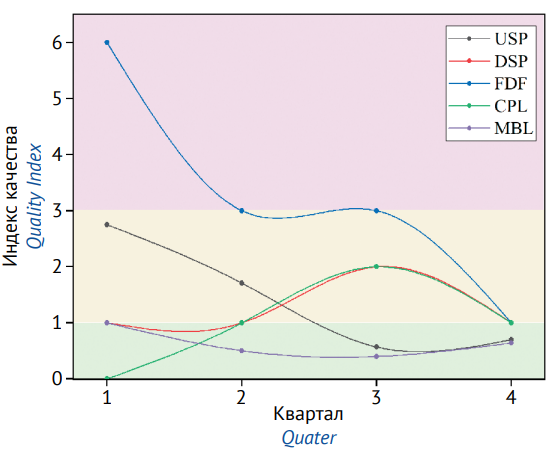
Рисунок подготовлен авторами по собственным данным / Figure is prepared by the authors using their own data
Рис. 2. Динамика изменения значений индекса качества (q) за год. USP — лаборатория культивирования; DSP — лаборатория выделения и очистки; FDF — лаборатория разработки готовых форм; CPL — физико-химическая лаборатория; MBL — микробиологическая лаборатория (цветом обозначены границы значений q)
Fig. 2. Time course of changes in the quality index (q) values throughout the studied year. USP, Upstream Process; DSP, Downstream Process; FDF, Finished Dosage Form; CPL, Chemical Physics Laboratory; MBL, Microbiological Laboratory (q-value limits are indicated with colours)
Предложена модель организации системы менеджмента качества научно-исследовательского центра фармацевтического предприятия по производству биотехнологических препаратов. Разработан универсальный механизм, позволяющий оценить эффективность функционирования системы менеджмента качества вне зависимости от индивидуальных требований к работе подразделений центра.
Введение индекса качества позволило регулярно осуществлять сбор оцифрованных данных и проводить унифицированный мониторинг системы менеджмента качества. На основании данного индекса были показаны тенденции улучшения системы менеджмента качества в подразделениях центра.
Следует отметить, что индекс качества может быть применен не только для оценки деятельности подразделений разработки фармацевтических лекарственных препаратов, но и перенесен на другие области разработки наукоемких продуктов ввиду его универсальности, простоты и возможности расширения его значений за счет введения иных показателей качества, важных для организации.
Кроме того, по нашим наблюдениям введение индекса качества способствует поддержанию здорового соревновательного духа между подразделениями и может являться дополнительным стимулом для сотрудников подразделений и их руководителей к совершенствованию системы менеджмента качества.
1 Current Good Manufacturing Practice (CGMP) Regulations. Food and Drug Administration; 2023.
2 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 77 «Об утверждении Правил надлежащей производственной практики Евразийского экономического союза».
EudraLex — Good Manufacturing Practice (GMP) guidelines. V. 4.
3 WHO Guidelines: Production. https://www.who.int/teams/health-product-and-policy-standards/standards-and-specifications/norms-and-standards-for-pharmaceuticals/guidelines/production
4 ICH Q7 Good Manufacturing Practice Guide for active pharmaceutical ingredients. CPMP/ICH/4106/00. London: EMA; 2000.
5 PE 009-16 Guide to Good Manufacturing Practice for Medicinal Products Annexes. PIC/S; 2022.
6 ICH Q8 (R2) Pharmaceutical development. EMA/CHMP/ICH/167068/2004. London: EMA; 2009.
7 Там же.
ICH Q11 Development and manufacture of drug substances (Chemical entities and biotechnological/biological entities).
EMA/CHMP/ICH/425213/2011. London: EMA; 2012.
8 QAS/20.865/.Rev1 Draft working document for comments: WHO good practices for research and development facilities of pharmaceutical products. WHO; 2021.
9 ISO/IEC 17025:2017. General requirements for the competence of testing and calibration laboratories.
ГОСТ ISO/IEC 17025-2019. Общие требования к компетентности испытательных и калибровочных лабораторий.
10 ENV/MC/CHEM(98)17. Principles on Good Laboratory Practice. OECD; 1997.
ГОСТ 33044-2014. Принципы надлежащей лабораторной практики.
11 Good Research Practice. Stockholm: Swedish Research Council; 2017.
UK Medical Research Council. https://www.ukri.org/about-us/mrc/our-policies-and-standards/research/
12 EMEA/CHMP/BMWP/101695/2006. Guideline on comparability of biotechnology-derived medicinal products after a change in the manufacturing process. London: EMA; 2007.
13 ICH Q8 (R2) Pharmaceutical development. EMA/CHMP/ICH/167068/2004. EMA; 2009.
14 ICH Q11 Development and manufacture of drug substances (Chemical entities and biotechnological/biological entities).
EMA/CHMP/ICH/425213/2011. EMA; 2012.
15 ГОСТ ISO/IEC 17025-2019. Общие требования к компетентности испытательных и калибровочных лабораторий.
16 Приказ Министерства экономического развития Российской Федерации от 26.10.2020 № 707 «Об утверждении критериев аккредитации и перечня документов, подтверждающих соответствие заявителя, аккредитованного лица критериям аккредитации».
17 ENV/MC/CHEM(98)17. Principles on Good Laboratory Practice. OECD; 1997.
ГОСТ 33044-2014. Принципы надлежащей лабораторной практики.
1. Fisher AC, Lee SL, Harris DP, Buhse L, Kozlowski S, Yu L, et al. Advancing pharmaceutical quality: An overview of science and research in the U.S. FDA’s Office of Pharmaceutical Quality. Int J Pharm. 2016;515(1–2):390–402. https://doi.org/10.1016/j.ijpharm.2016.10.038
2. Li TW, Tu PW, Liu LL, Wu SI. Assurance of medical device quality with quality management system: an analysis of good manufacturing practice implementation in Taiwan. Biomed Res Int. 2015;2015:670420. https://doi.org/10.1155/2015/670420
3. Ravinetto R, Roosen T, Dujardin C. The Belgian commitment to pharmaceutical quality: a model policy to improve quality assurance of medicines available through humanitarian and development programs. J Pharm Policy Pract. 2018;11:12. https://doi.org/10.1186/s40545-018-0136-z
4. Jampilek J, Crowley PJ, Olsen M, Tam K. Modern approaches to quality assurance of drug formulations. Biomed Res Int. 2015;2015:126478. https://doi.org/10.1155/2015/126478
5. Bereda G. Quality assurance and quality control. Current furtherances and hereafter point of view. J Anal Pharm Res. 2021;10(5):212–5. https://doi.org/10.15406/japlr.2021.10.00386
6. Kushare S, Darekar A, Saudagar R. Quality assurance and quality management in pharmaceutical science and pharmaceutical industry. J Drug Deliv Ther. 2019;9(2-S):537–42.
7. Haleem RM, Salem MY, Fatahallah FA, Abdelfattah LE. Quality in the pharmaceutical industry — A literature review. Saudi Pharm J. 2015;23(5):463–69. https://doi.org/10.1016/j.jsps.2013.11.004
8. Yu LX, Amidon G, Khan MA, Hoag SW, Polli J, Raju GK, Woodcock J. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–83. https://doi.org/10.1208/s12248-014-9598-3
9. Lee SH, Kim JK, Jee JP, Jang DJ, Park YJ, Kim JE. Quality by Design (QbD) application for the pharmaceutical development process. J Pharm Investig. 2022;52:649–82. https://doi.org/10.1007/s40005-022-00575-x
10. Zagalo DM, Silva BMA, Silva C, Simões S, Sousa JJ. A quality by design (QbD) approach in pharmaceutical development of lipid-based nanosystems: A systematic review. J Drug Deliv Sci Technol. 2022;70:103207. https://doi.org/10.1016/j.jddst.2022.103207
11. Testas M, Sais Tda C, Medinilha LP, Niwa KNI, de Carvalho LS, Maia SD, et al. An industrial case study: QbD to accelerate time-to-market of a drug product. AAPS Open. 2021;7:12. https://doi.org/10.1186/s41120-021-00047-w
12. Weitzel J, Pappa H, Banik GM, Barker AR, Bladen E, Chirmule N, et al. Understanding quality paradigm shifts in the evolving pharmaceutical landscape: perspectives from the USP quality advisory group. AAPS J. 2021;23(6):112. https://doi.org/10.1208/s12248-021-00634-5
13. Mantus D, Pisano DJ, eds. FDA Regulatory Affairs. London: CRC Press; 2014. https://doi.org/10.1201/b16471
14. Patravale VB, Disouza JI, Rustomjee M, eds. Pharmaceutical product development. Insights into pharmaceutical processes, management and regulatory affairs. Boca Raton: CRC Press; 2016. https://doi.org/10.1201/b19579
Гиба Иван Сергеевич, канд. физ.-мат. наук
Ул. Связи, д. 34а, Санкт-Петербург, пос. Стрельна, 198515
Салиева Камилла Рахмановна
Ул. Связи, д. 34а, Санкт-Петербург, пос. Стрельна, 198515
Батуева Анастасия Александровна
Ул. Связи, д. 34а, Санкт-Петербург, пос. Стрельна, 198515
Григорьева Ирина Владимировна
Ул. Связи, д. 34а, Санкт-Петербург, пос. Стрельна, 198515
Драй Роман Васильевич, канд. мед. наук
Ул. Связи, д. 34а, Санкт-Петербург, пос. Стрельна, 198515
Гиба И.С., Салиева К.Р., Батуева А.А., Григорьева И.В., Драй Р.В. Внедрение и оценка системы менеджмента качества в области фармацевтической разработки биотехнологических лекарственных препаратов. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):62-71. https://doi.org/10.30895/1991-2919-2023-549
Giba I.S., Salieva K.R., Batueva A.A., Grigorieva I.V., Drai R.V. Quality Management in Pharmaceutical Development of Biotechnology-Derived Medicinal Products: Implementation and Evaluation. Bulletin of the Scientific Centre for Expert Evaluation of Medicinal Products. Regulatory Research and Medicine Evaluation. 2024;14(1):62-71. (In Russ.) https://doi.org/10.30895/1991-2919-2023-549

Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных




























