



«Регуляторные исследования и экспертиза лекарственных средств» – научно-практический рецензируемый журнал открытого доступа, выпускаемый в печатной и онлайн-версиях. Основан в 1999 году.
Цель журнала: содействие развитию фармацевтической и медицинской науки и практики посредством опубликования и распространения информации о передовых достижениях в регуляторной деятельности и сфере обращения лекарственных средств.
Целевая аудитория. Предназначен для широкого круга специалистов, как российских, так и зарубежных, работающих в сфере обращения лекарственных средств:
- разработчиков и производителей лекарственных препаратов;
- представителей экспертных организаций, государственных регуляторных органов;
- работников контрольно-разрешительной системы и государственного надзора в сфере обращения лекарственных средств;
- сотрудников научно-исследовательских институтов, преподавателей, аспирантов и студентов медицинских, фармацевтических вузов, врачей и провизоров.
Более подробная информация – в разделе Цели и задачи.
Учредитель: ФГБУ «Научный центр экспертизы средств медицинского применения» Министерства здравоохранения Российской Федерации.
Периодичность: 6 раз в год.
Импакт-фактор: двухлетний импакт-фактор РИНЦ (2024) – 0,533.
Редколлегия. Географическое представительство:
- 8 стран
- 12 городов
Рецензирование:
- Двойное слепое
- Минимум 2 рецензента на рукопись
Основные метрики журнала:
14 дней в среднем от подачи до первого решения
88 дней в среднем от подачи до публикации в Интернете
15% приглашенных авторов
68% доля принятия рукописей
64 тыс. загрузок PDF в 2025 г.
Плата за публикацию: бесплатно.
Индексация. Индексируется в российских и международных реферативных и полнотекстовых базах, включен в наукометрические базы данных РИНЦ, RSCI, входит в "Белый список" научных изданий и Перечень ВАК (категория К1).
Информация об индексации в других российских и международных базах доступна в разделе Индексирование.
Регистрация. Свидетельство о регистрации средства массовой информации ПИ № ФС77-82931 от 14 марта 2022 г.
Подписка. Подписной индекс в каталоге Пресса России – 57942, Урал-Пресс – 57942.
Текущий выпуск
ГЛАВНАЯ ТЕМА: РЕГУЛИРОВАНИЕ ОБРАЩЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ В ЕАЭС: РЕГИСТРАЦИЯ И КОНТРОЛЬ КАЧЕСТВА

31.12.2025 заканчивается переход к правилам регулирования обращения лекарственных средств в рамках Евразийского экономического союза (ЕАЭС). Как подчеркивает начальник контрольно-организационного управления ФГБУ «НЦЭСМП» Минздрава России Екатерина Михайловна Рычихина, завершение переходного периода — это не финишная черта, а скорее новый виток развития. В эксклюзивном интервью эксперт с уникальным опытом практической работы выявляет скрытые риски, которые могут возникнуть даже после успешного приведения досье в соответствие, подчеркивает критические аспекты гармонизации, требующие особого внимания регуляторных органов и производителей. Особое внимание в беседе уделяется последним изменениям в правилах обращения лекарственных средств на территории ЕАЭС, которые уже сегодня формируют новую правовую реальность. В интервью не только раскрывается официальная позиция учреждения, но и даются практические рекомендации, которые помогут компаниям избежать ошибок и выстроить эффективную стратегию взаимодействия с экспертной организацией.
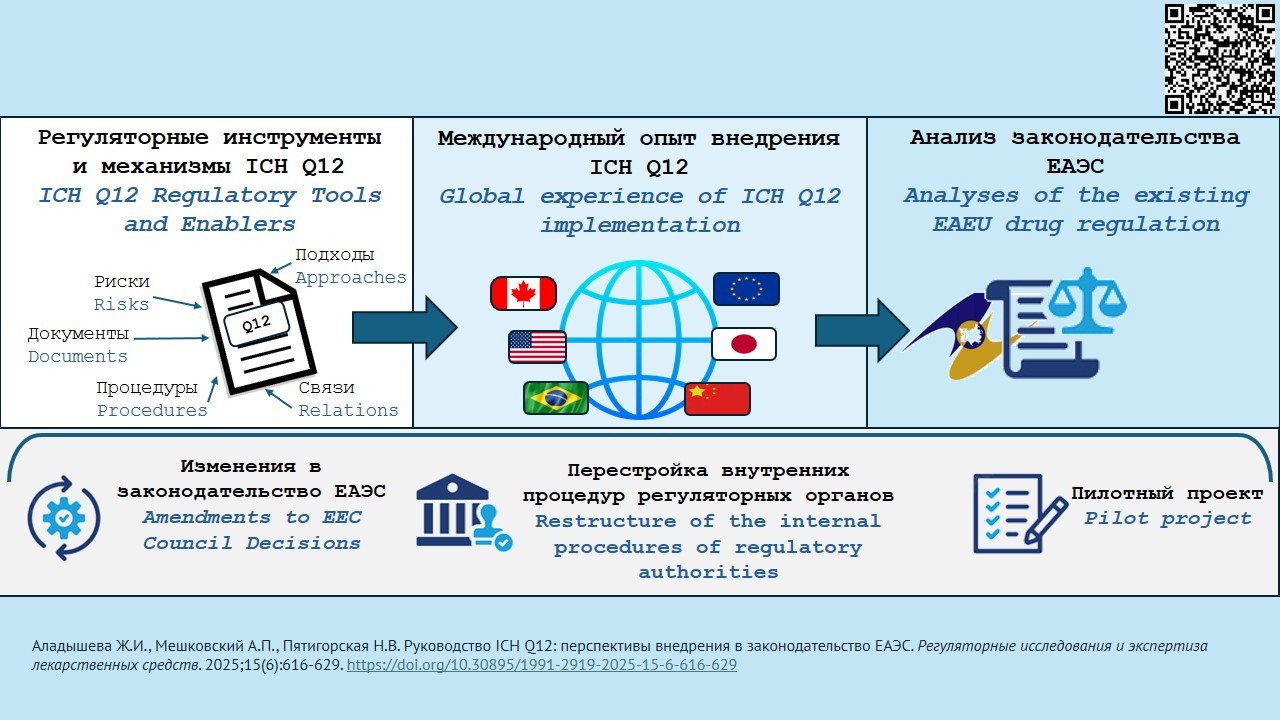
ВВЕДЕНИЕ. В 2019 г. Международный совет по гармонизации технических требований к лекарственным средствам (ICH) принял Руководство ICH Q12 по управлению жизненным циклом фармацевтического продукта. Оно охватывает все категории изменений в Модуль 3 «Качество» регистрационного досье на лекарственный препарат и в мастер-файлы на фармацевтическую субстанцию и другие продукты. Ожидается, что его применение поможет оптимизировать управление изменениями, уменьшить время их согласования и реализации, снизить риски для пациентов. По состоянию на 01.08.2025 документ внедрен в трех странах-участницах ICH: США, Японии и КНР.
ЦЕЛЬ. Оценка перспективы внедрения Руководства ICH Q12 в законодательство ЕАЭС.
ОБСУЖДЕНИЕ. Рассмотрены 8 инструментов и механизмов, рекомендуемых к использованию при управлении пострегистрационными изменениями в области качества, оценена степень их новизны по отношению к общепринятым принципам и подходам к регулированию качества лекарственных средств. Проведен анализ опыта стран-участниц ICH, внедривших или внедряющих данное руководство, проведенных пилотных проектов и сопутствующих внедрению изменений законодательства и внутренних процедур. На данный момент полноценное внедрение документа было осуществлено только в США. Проанализирован опыт фармацевтических компаний, вносивших изменения согласно ICH Q12. Проведен анализ действующего законодательства ЕАЭС в части регулирования лекарственных средств, и установлено, что для эффективного использования регуляторных инструментов и механизмов Руководства ICH Q12 необходимо внести ряд принципиальных изменений в Решения Совета ЕЭК № 77, 78, 84, 91, издать адаптированное к законодательству ЕАЭС Руководство Q12, провести перестройку внутренних процедур регуляторных органов и их подведомственных экспертных учреждений и запустить пилотный проект по тестированию новых административных процедур.
ВЫВОДЫ. Внедрение Руководства Q12 позволит трансформировать внесение изменений в области качества в зарегистрированные лекарственные препараты из сложного административного барьера в механизм инноваций, будет содействовать научно-техническому прогрессу и постоянному улучшению продукции. Для более широкого внедрения руководства потребуется изменение законодательства, а также изменение внутренних процедур регуляторных органов и фармацевтических компаний. Из предложенных механизмов Q12 наиболее успешно проходит внедрение протоколов управления изменениями. В ЕАЭС имеются все основные условия для внедрения Q12; однако при этом требуется решение ряда принципиальных и технических вопросов, которые затрагивают национальные системы регулирования лекарственных средств государств – участников ЕАЭС, изменение имеющихся документов и разработка новых.
ЦИФРОВЫЕ ТЕХНОЛОГИИ В ФАРМАЦИИ

ВВЕДЕНИЕ. Существующие методы подготовки документов, используемые в ходе разработки лекарственных средств, характеризуются высокими временными затратами (40–60% рабочего времени специалистов), высокой частотой ошибок, вносимых в документацию, и ограниченной интероперабельностью данных. Увеличение эффективности подготовки документов возможно при использовании нейросетевых технологий и переходе к комплексной автоматизации процедур жизненного цикла регистрационного досье.
ЦЕЛЬ. Оценка возможности использования систем искусственного интеллекта (ИИ) и машинного анализа при подготовке регистрационного досье лекарственного препарата в процессе разработки лекарственного средства.
ОБСУЖДЕНИЕ. Модели обработки естественного языка (NLP) показывают высокую эффективность в области обработки технической и регуляторной документации. Системы распознавания именованных сущностей (NER) с точностью извлечения 89–96% позволяют сократить время обработки (подготовки и последующей проверки) производителем материалов при формировании электронного общего технического документа на 64%, однако возможность обработки информации ограничена трудностями интерпретации морфологически сложных терминов и требует использования наборов аннотированных данных. Следует отметить, что генеративные модели типа GPT-4 без дополнительной настройки при использовании в архитектуре генерации, дополненной поиском (RAG), могут формировать фактологически некорректную информацию. Предиктивные системы на основе графовых нейросетей и ансамблей XGBoost демонстрируют высокую точность (ROC AUC до 0,88) при прогнозировании одобрения препаратов, но имеют недостаток в виде невозможности интерпретации решений и систематических смещений в данных. Разработка документоцентричных платформ с NLP позволяет сократить время подготовки досье на 60%, однако при внедрении автоматизированной процедуры формирования разделов досье требуется экспертная верификация.
ВЫВОДЫ. Концепция интегрированных ИИ-систем подтверждает свою эффективность, сокращая сроки обработки документов производителем и повышая точность решений, что способствует ускорению вывода препаратов на рынок. Перспективы внедрения цифровых технологий связаны с преодолением различий в терминах через унифицированные онтологии. Для практической реализации требуется разработка единых стандартов валидации ИИ-алгоритмов и адаптивных систем.
РАЗРАБОТКА ЛЕКАРСТВЕННЫХ СРЕДСТВ
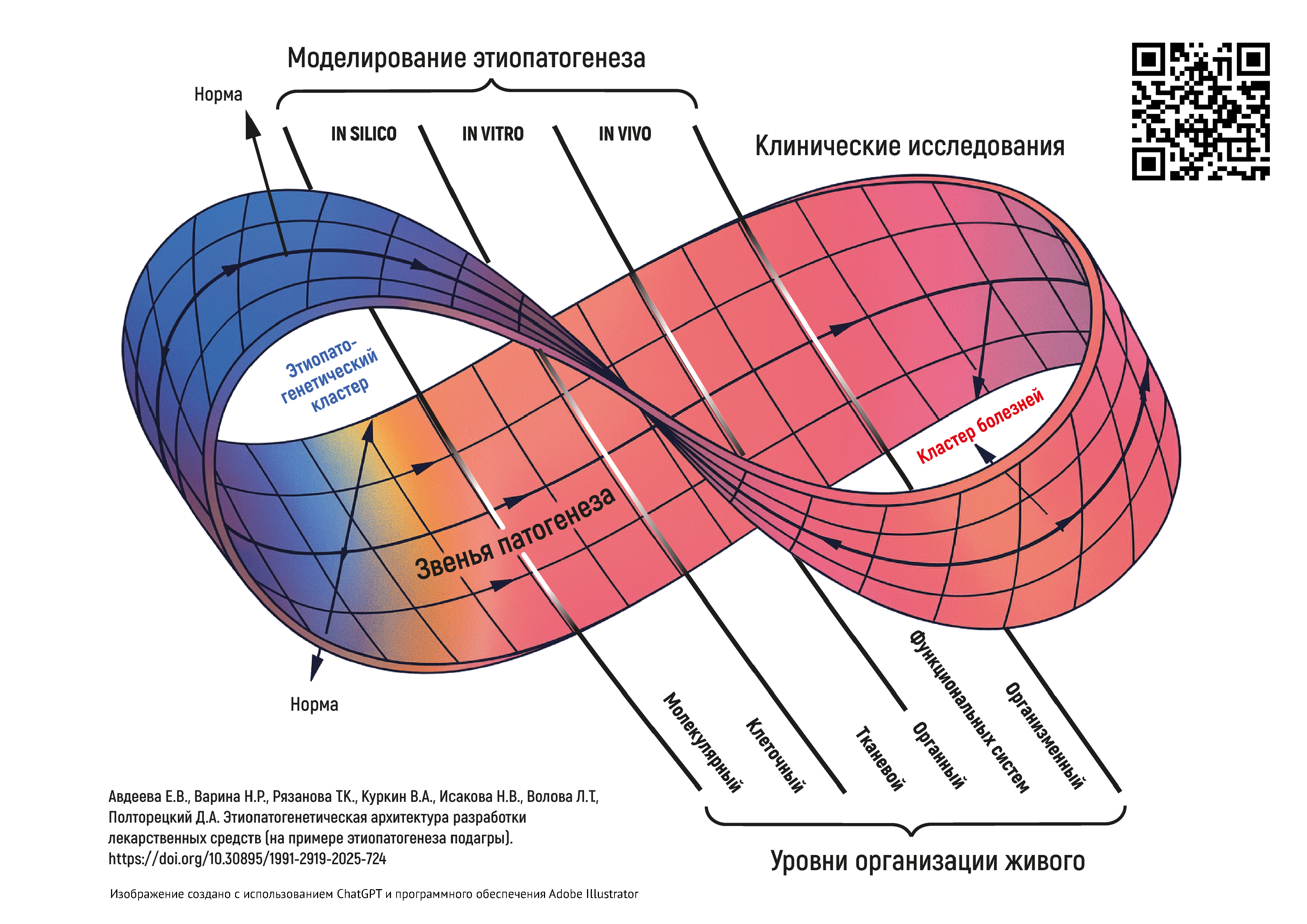
ВВЕДЕНИЕ. Системное понимание патофизиологических и патобиохимических путей заболеваний и патологических состояний является насущной потребностью как для врачей при постановке диагноза и лечении конкретного пациента, так и для разработчиков лекарственных средств (ЛС). Накопленные знания в медицинской и в смежных сферах, быстрое развитие цифровых инструментов делают возможным на качественно новом уровне смоделировать систему реактивности организма в норме и патологии. Это приведет к созданию цифровой архитектуры состояний организма с взаимосвязанными звеньями патогенеза, которые и будут находиться в фокусе внимания исследователей от дизайна ранней разработки ЛС до клинических исследований.
ЦЕЛЬ. Ревизия имеющихся подходов и построение на их основе этиопатогенетической архитектуры состояний и заболеваний как фундаментальной основы целенаправленной разработки лекарственных средств.
ОБСУЖДЕНИЕ. В исследовании были показаны необходимость и возможность (на примере подагры) построения объемной этиопатогенетической архитектуры состояний и заболеваний организма, основанной на иерархических связях патологических процессов на разных уровнях организации живого. Обоснованы основные векторы ее использования: для медицинских целей — основанная на данных диагностика состояний и заболеваний индивида, персонификация фармакотерапии; для фармацевтических целей — основа для исследования фармакодинамики ЛС начиная со скрининга веществ-кандидатов, использования методологических возможностей таргетных и мультитаргетных подходов в разработке ЛС. На примере архитектуры этиопатогенеза подагры обсуждена логика в разработке дизайна исследования ЛС.
ВЫВОДЫ. Предложена методология построения этиопатогенетической архитектуры как отражение причинно-следственных связей с разным уровнем значимости в формировании патологических состояний и заболеваний организма. Этиопатогенетический подход должен стать связующей основой между всеми этапами создания и применения новых ЛС, а также при исследовании возможности расширения показаний к применению уже использующихся ЛС. Появляются новые возможности и для разработки этиопатогенетических моделей разных уровней сложности: от drug-дизайна на молекулярном уровне до моделирования патофизиологических процессов на организменном уровне.
КОНТРОЛЬ КАЧЕСТВА
ВВЕДЕНИЕ. Эффективное функционирование системы контроля качества лекарственных препаратов невозможно без использования стандартных образцов в процессе валидации и верификации аналитических методик. Особую актуальность приобретает разработка стандартных образцов для контроля качества фармацевтических субстанций, впоследствии использующихся в целях фармацевтической разработки новых лекарственных препаратов.
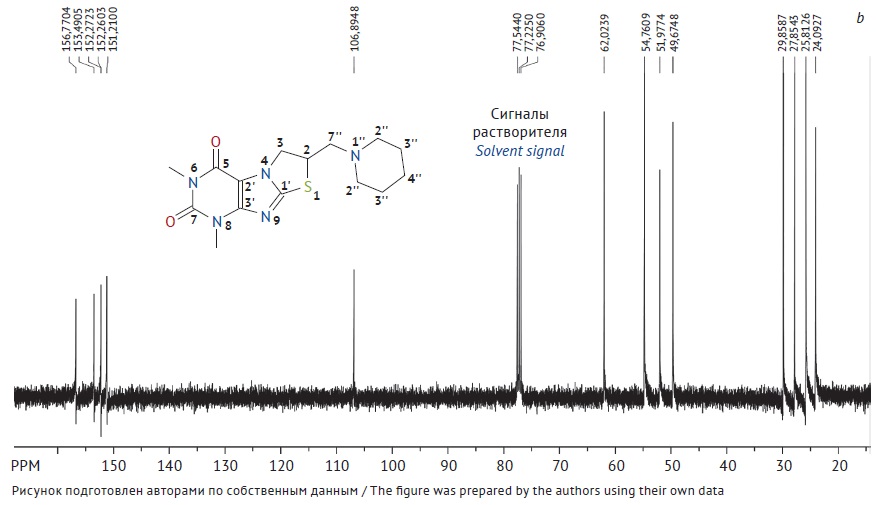
ЦЕЛЬ. Разработка способа получения и проведение аттестации первичного стандартного образца 6,8-диметил-2-пиперидинометил-2,3-дигидротиазоло[2,3-F]ксантина для его последующего применения в контроле качества лекарственных средств.
МАТЕРИАЛЫ И МЕТОДЫ. Стандартный образец получали перекристаллизацией
6,8-диметил-2-пиперидинометил-2,3-дигидротиазоло[2,3-F]ксантина из этанола. Структуру стандартного образца устанавливали методами ядерного магнитного резонанса (ЯМР) и инфракрасной (ИК) спектроскопии, чистоту определяли методом баланса масс, методом высокоэффективной жидкостной хроматографии (родственные примеси), а также методом неводной ацидиметрии.
РЕЗУЛЬТАТЫ. Выполнена оценка физико-химических характеристик стандартного образца 6,8-диметил-2-пиперидинометил-2,3-дигидротиазоло[2,3-F]ксантина, в том числе подтверждение структуры методами ИК- и ЯМР-спектроскопии, определение потери в массе при высушивании 0,073±0,015%, сульфатной золы 0,080±0,009%, родственных примесей (не обнаружено), тяжелых металлов (не более 0,002%) и элементного состава (С — 53,68±0,17%; Н — 6,32±0,02%; N — 20,81±0,09%; O — 9,52±0,06%; S — 9,57±0,04%), количественное определение методом неводной ацидиметрии 99,74±0,12% и методом баланса масс 99,85±0,01%.
ВЫВОДЫ. Предложен способ получения стандартного образца 6,8-диметил- 2-пиперидинометил-2,3-дигидротиазоло[2,3-F]ксантина. Физико-химические характеристики полученного образца соответствуют требованиям, предъявляемым к стандартному образцу, что позволяет рекомендовать данный образец для использования в качестве эталонного материала при проведении контроля качества лекарственных средств 6,8-диметил-2-пиперидинометил2,3-дигидротиазоло[2,3-F]ксантина.
ВВЕДЕНИЕ. Генистеин (5,7-дигидрокси-3-(4-гидроксифенил)хромен-4-он) — полифенольное соединение, обладающее выраженной антиоксидантной активностью и низкой токсичностью. Генистеин может быть применен для профилактики и лечения радиационных поражений при химиолучевой терапии. Однако включение в терапевтические схемы в качестве противорадиационного средства возможно только после его регистрации в качестве лекарственного средства. Для этого необходимо в том числе разработать методики оценки содержания генистеина, соответствующие фармакопейным требованиям к аналитическим методикам.
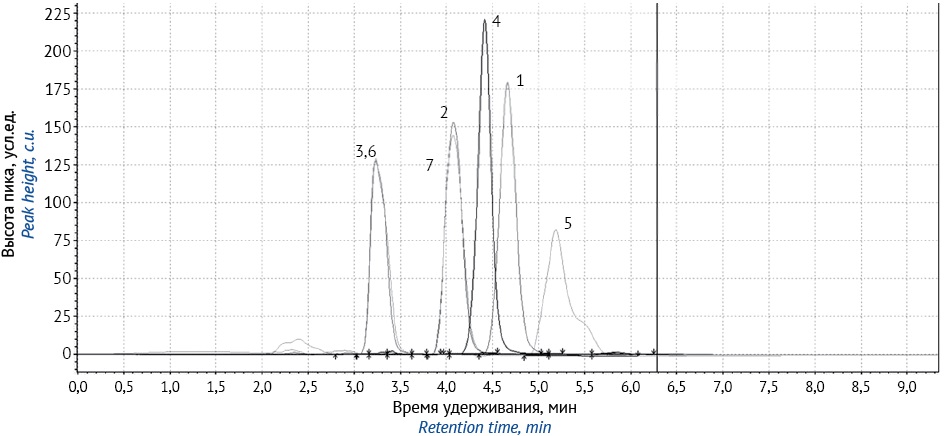
ЦЕЛЬ. Разработка методики идентификации и количественного определения потенциально активной фармацевтической субстанции генистеина методом высокоэффективной жидкостной хроматографии.
МАТЕРИАЛЫ И МЕТОДЫ. Образцы фармацевтической субстанции генистеина, синтезированные в ФГБОУ ВО СПХФУ Минздрава России, проанализированы с помощью системы высокоэффективной жидкостной хроматографии с диодно-матричным детектированием (ВЭЖХ-ДМД) Prominence LC-20 Shimadzu. Температура в отсеке для колонки — 40 °C, объем инжекции — 100 мкл, длина волны детекции — 261 нм, колонка Shimadzu C18 (4,6 мм×250 см, размер частиц 5 мкм).
РЕЗУЛЬТАТЫ. Разработана методика качественного и количественного определения генистеина методом ВЭЖХ-ДМД, подвижная фаза ацетонитрил : вода очищенная (75:25 об.:об., скорость потока — 0,7 мл/мин). Проведена оценка пригодности разработанной системы. Методика валидирована по показателям: специфичность, линейность (0,001–0,01%; r=0,997), правильность (RSD=1,26%), воспроизводимость (RSD=0,43%), внутридневная прецизионность (RSD<2%) и робастность.
ВЫВОДЫ. Разработанная методика качественного и количественного определения генистеина методом ВЭЖХ-ДМД соответствует фармакопейным требованиям к аналитическим методикам и может быть предложена для включения в проект фармакопейной статьи «Генистеин».

ВВЕДЕНИЕ. Бета-глюканы и пептидогликаны — компоненты клеточных стенок бактерий и грибов, которые могут являться источниками пирогенных загрязнений лекарственных препаратов парентерального применения. Присутствие подобных примесей может приводить к неблагоприятным иммунным реакциям, поэтому контролю присутствия бета-глюканов и пептидогликанов в лекарственных средствах уделяют определенное внимание. Унифицированный подход к обнаружению бета-глюканов и (или) пептидогликанов в фармацевтической отрасли отсутствует; практическое применение имеют несколько методов качественного и количественного определения этих примесей.
ЦЕЛЬ. Оценка практической значимости существующих методов обнаружения бета-глюканов и (или) пептидогликанов в лекарственных препаратах.
МАТЕРИАЛЫ И МЕТОДЫ. Исследована возможность применения методов определения бета-глюканов и пептидогликанов с использованием реактива на основе лизата амебоцитов и реактива из плазмы личинок шелкопряда. Содержание бета-глюканов определяли в лекарственном препарате «Бупивакаин», в котором бета-глюканы были обнаружены ранее случайным образом. В испытаниях препарата использовали три типа реактивов на основе лизата амебоцитов разного состава, реагирующих: на бактериальные эндотоксины и бета-глюканы (лизат с факторами С и G); только на бактериальные эндотоксины (лизат с фактором С); только на бета-глюканы (лизат с фактором G). Наличие пептидогликанов оценивали в лекарственном препарате «Икодекстрин» с помощью реактива из плазмы личинок шелкопряда. Качественный анализ выполняли путем визуальной оценки окраски испытуемых растворов после нагревания их в суховоздушном термоблоке. Количественное определение проводили кинетическим фотоколориметрическим методом, обработку первичных данных выполняли с использованием программной среды R.
РЕЗУЛЬТАТЫ. В результате испытаний с реактивами лизата амебоцитов (факторы С и G) и (фактор С) определено присутствие бета-глюканов в препарате «Бупивакаин»; количественное содержание бета-глюканов (более 2000 пг/мл) определено хромогенным кинетическим методом с помощью лизата амебоцитов (фактор G). В препарате «Икодекстрин» превышения нормативного содержания пептидогликанов (не более 200 пг/мл) не зафиксировано.
ВЫВОДЫ. Методы определения бета-глюканов и (или) пептидогликанов с использованием лизата амебоцитов и реактива из плазмы личинок шелкопряда могут быть применены для выявления данных примесей в лекарственных препаратах. При определении метода исследования следует учитывать состав лекарственного препарата и цель определения примесей.
ЛЕКАРСТВЕННЫЕ СРЕДСТВА РАСТИТЕЛЬНОГО ПРОИСХОЖДЕНИЯ

ВВЕДЕНИЕ. Экстракционные растительные препараты широко используются в медицинской практике. Одним из показателей качества этих препаратов является содержание элементных примесей. К числу факторов, влияющих на состав продукта, относится технология его изготовления. Данные об особенностях перехода элементов, в том числе тяжелых металлов и мышьяка, из исходного растительного сырья в экстракционные лекарственные формы практически отсутствуют.
ЦЕЛЬ. Оценка перехода элементных примесей из лекарственного растительного сырья в настойки, изготовленные различными методами экстракции.
МАТЕРИАЛЫ И МЕТОДЫ. Объект исследования — трава пустырника, использующаяся для промышленного производства настоек (АО «Флора Кавказа»). В лабораторных условиях из нее были изготовлены настойки методами дробной мацерации, ультразвуковой экстракции и вихревой экстракции. Элементный состав исходного растительного сырья и полученных настоек определен методом масс-спектрометрии с индуктивно-связанной плазмой на приборе Agilent ICP MS 7900.
РЕЗУЛЬТАТЫ. В траве пустырника и полученных из нее настойках определено 13 элементов (V, Cr, Mn, Fe, Co, Ni, Cu, Zn, As, Sr, Cd, Tl, Pb), Hg не обнаружена. Концентрации элементов в исходном сырье составили 0,007–121,098 мг/кг, содержание токсичных элементов (Pb, Cd, As) соответствовало фармакопейным требованиям. В настойках содержание тяжелых металлов не превышало 1,25 мг/кг, в бóльших концентрациях присутствовали Zn, Cu, Mn, в минимальных — Tl и Cd. Проведена оценка возможного поступления элементов в организм человека с изученными извлечениями и безопасности их медицинского применения. Рассчитаны степени перехода тяжелых металлов и мышьяка из исходного растительного сырья в полученные настойки — для большинства элементов они не превышали 46%. Установлено, что в наибольших количествах во все изученные настойки извлекается Zn, в наименьших — Cd. При использовании дробной мацерации в наибольших количествах в настойки переходили Zn, Ni, Cu и Tl, вихревой экстракции — V, Cr, Co и Sr. При использовании ультразвуковой экстракции в раствор переходит наименьшее количество примесей тяжелых металлов.
ВЫВОДЫ. Изучен переход элементов в спиртоводные извлечения при разных методах экстракции на примере настоек пустырника. Установлено, что в случае использования методов дробной мацерации и вихревой экстракции могут быть получены настойки с более высоким содержанием эссенциальных элементов, а при использовании ультразвуковой экстракции — с минимальными концентрациями токсичных примесей тяжелых металлов (Pb, Cd) и As.
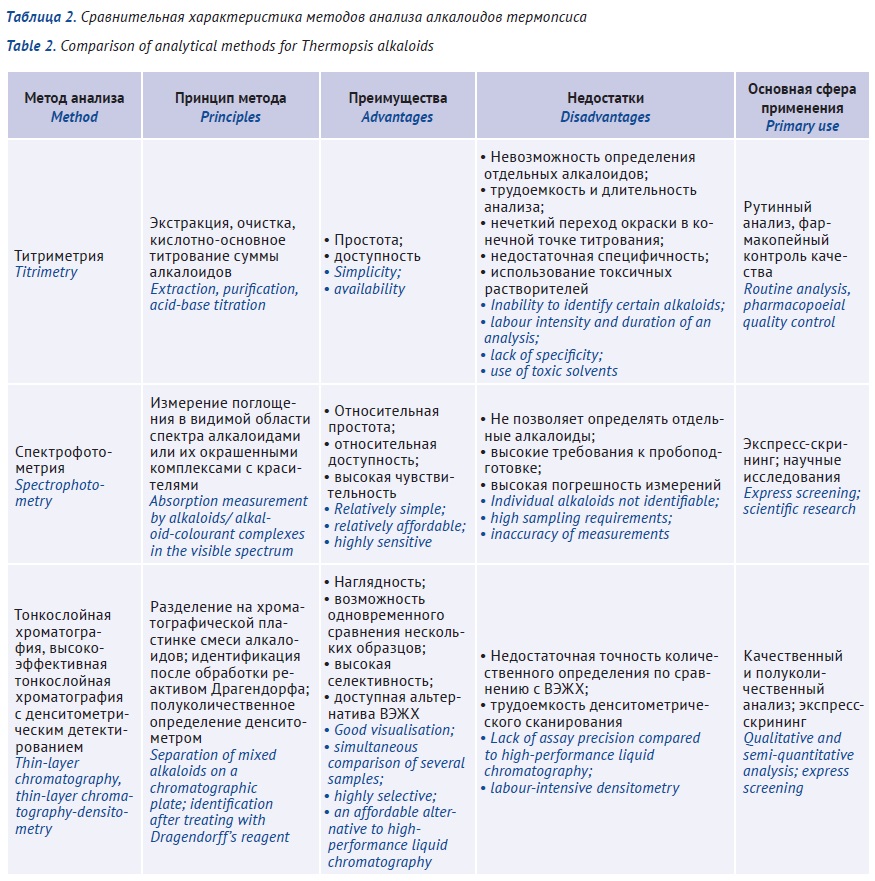
ВВЕДЕНИЕ. Основными алкалоидами травы термопсиса ланцетного являются термопсин (N-метилцитизин), пахикарпин и анагирин; в семенах преобладает цитизин, применяемый для лечения табачной зависимости. Различия в токсичности и фармакологической активности индивидуальных алкалоидов требуют не только контроля их общей суммы, но и мониторинга содержания каждого из них.
ЦЕЛЬ. Оценка перспективности применения отдельных аналитических методов для количественного определения алкалоидов термопсиса.
ОБСУЖДЕНИЕ. Фармакопейный титриметрический метод является трудоемким, длительным, недостаточно точным, поскольку определение конечной точки титрования сопряжено с существенной погрешностью; требует использования токсичных растворителей и может быть использован только для определения суммы алкалоидов без их дифференциации. Спектрофотометрические методы не позволяют надежно определять индивидуальные алкалоиды из-за недостаточной селективности (наложение спектров отдельных алкалоидов), результаты чувствительны к условиям пробоподготовки. Наиболее надежными инструментальными методами для качественного и количественного анализа травы термопсиса являются тонкослойная хроматография и высокоэффективная жидкостная хроматография.
ВЫВОДЫ. В фармакопейном анализе для разделения и количественного определения индивидуальных алкалоидов (идентификации цитизина и термопсина), а также для идентификации примесей семян термопсиса в лекарственных препаратах, не предназначенных для лечения табачной зависимости, могут быть использованы методы тонкослойной и высокоэффективной жидкостной хроматографии.
ВВЕДЕНИЕ. Растения семейства бурачниковые (Boraginaceae) Pulmonaria mollis, Nonea rossica, Onosma simplicissima и Cynoglossum officinale широко распространены на территории Российской Федерации и могут являться источниками лекарственных средств, обладающих противомикробными, противоанемическими, антикоагулянтными свойствами. В настоящее время данные растения не являются официнальными, что
может быть связано с содержанием в них пирролизидиновых алкалоидов, способных вызывать гепатотоксические эффекты.
ЦЕЛЬ. Оценка безопасности применения растений семейства Boraginaceae по критерию острой токсичности и содержанию пирролизидиновых алкалоидов.
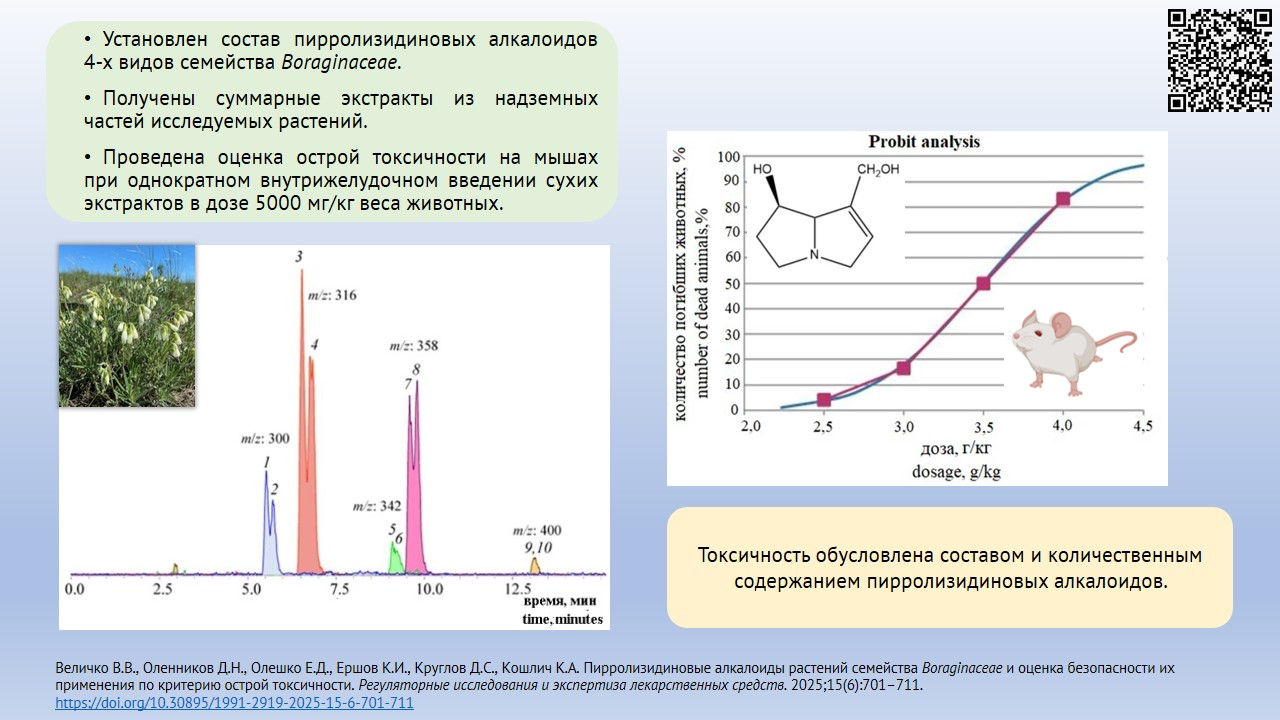
МАТЕРИАЛЫ И МЕТОДЫ. В качестве объектов исследования использовали высушенные надземные части растений Pulmonaria mollis, Nonea rossica, Onosma simplicissima и Cynoglossum officinale, заготовленные в период цветения. Состав и содержание алкалоидов в спиртовых извлечениях определяли методом высокоэффективной жидкостной хроматографии с диодно-матричным и масс-спектрометрическим детектированием с ионизацией электрораспылением. Определение острой токсичности проводили на 102 половозрелых мышах обоего пола стока CD-1 весом 24,0±2,0 г в возрасте 12 нед., полученных из ЦКП «Виварий конвенциональных животных» ФГБНУ ФИЦ ИЦиГ СО РАН. Животным однократно внутрижелудочно вводили сухие экстракты изучаемых видов сырья, растворенные в дистиллированной воде, в дозе 5 г/кг.
РЕЗУЛЬТАТЫ. Установлено отсутствие пирролизидиновых алкалоидов в траве P. mollis, наличие их следовых количеств (0,01 мкг/г) в листьях P. mollis. В других изученных видах растений обнаружены пирролизидиновые алкалоиды — энантиомеры интермедина и ликопсамина и их производные: в траве O. simplicissima — 1,07±0,03 мкг/г, в траве N. rossica — 8,25±0,08 мкг/г, в траве C. officinale — 676,3±7,4 мкг/г. По результатам оценки острой токсичности сухие экстракты из травы и листьев P. mollis, травы N. rossica и травы O. simplicissima отнесены к 5 классу токсичности, экстракт из травы C. officinale — к 4 классу токсичности.
ВЫВОДЫ. Извлечения из травы и листьев P. mollis в исследуемых дозах являются нетоксичными, для извлечений из травы O. simplicissima и N. rossica необходимо проведение дальнейших исследований для определения токсичности при длительном применении. Извлечения из травы C. officinale токсичны и не могут использоваться для внутреннего применения.
АПТЕЧНОЕ ИЗГОТОВЛЕНИЕ
ВВЕДЕНИЕ. На современном фармацевтическом рынке ассортимент готовых жидких лекарственных форм поливитаминов для детей от 1 года до 3 лет представлен в основном биологически активными добавками. Они содержат вспомогательные вещества (консерванты, сахарозу, синтетические ароматизаторы и красители), которые небезопасны для применения у детей младшего возраста. Экстемпоральное изготовление дает возможность минимизировать количество вспомогательных веществ, нежелательных для детского организма. Перспективными путями усовершенствования экстемпорального мелкосерийного изготовления являются: унификация прописей, модернизация технологического процесса и экспериментально обоснованное продление сроков годности лекарственного препарата.
ЦЕЛЬ. Подбор состава и оптимизация технологии поливитаминного сиропа аптечного изготовления для педиатрической практики.
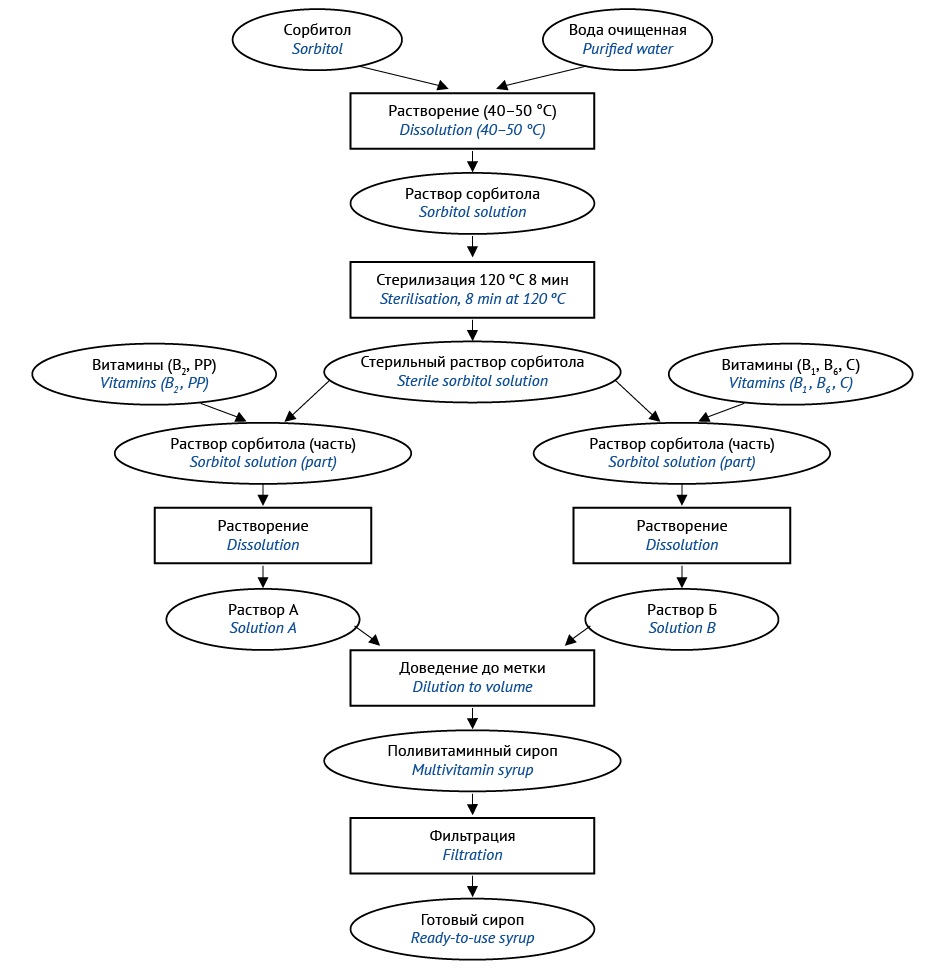
МАТЕРИАЛЫ И МЕТОДЫ. Объект исследования: сорбитолсодержащий поливитаминный сироп аптечного изготовления, в состав которого входят водорастворимые витамины: аскорбиновая кислота, никотиновая кислота, тиамина гидрохлорид, рибофлавин, пиридоксина гидрохлорид (С, РР, В1, В2, В6) в концентрациях, соответствующих физиологическим потребностям детей младшего возраста от 1 года до 3 лет. Для получения сиропа использовали фармацевтические субстанции, соответствующие требованиям нормативной документации на вышеуказанные витамины и сорбитол. Компоненты идентифицированы специфическими качественными реакциями, количественное содержание компонентов определяли химическими и физико-химическими методами (спектрофотометрия, спектрофлуориметрия, рефрактометрия), модифицированными с учетом совместного присутствия витаминов в составе сиропа. В ходе анализа контролировали технологические (плотность, значение рН) и органолептические (вкус, запах, цвет) показатели сиропа.
РЕЗУЛЬТАТЫ. Для изготовления поливитаминного сиропа в условиях производственных аптек предложены два варианта технологии: с использованием стерильного раствора сорбитола (технология 1) и с применением для стерилизации метода мембранной фильтрации готового продукта (технология 2). Проведен качественный и количественный анализ сиропов, полученных обоими методами. Показано, что технологические (плотность, значение рН) и органолептические показатели (цвет, запах, вкус) не изменились в течение 30 сут при хранении в защищенном от света прохладном месте.
ВЫВОДЫ. Предложены два варианта технологии изготовления сорбитолсодержащего сиропа, в состав которого включены водорастворимые витамины С, РР, В1, В2, В6 в концентрациях, соответствующих физиологическим потребностям детей в возрасте от 1 года до 3 лет. Обе предлагаемые технологии могут быть использованы в условиях производственных аптек, однако применение мембранной фильтрации повышает себестоимость изготовления, поскольку требует наличия дополнительного лабораторного оборудования (система вакуумной фильтрации и вакуумный насос).
Объявления
2025-06-03
Поздравляем с избранием в академики РАН!
Редколлегия и редакция журнала «Регуляторные исследования и экспертиза лекарственных» поздравляют члена редколлегии – Андрея Дмитриевича Дурнева, избранного в действительные члены Российской академии наук.
| Еще объявления... |

ISSN 3034-3453 (Online)





























