




Содержание
Перейти к:
https://doi.org/10.30895/1991-2919-2023-537
Перейти к:
АКТУАЛЬНОСТЬ. Биовейвер — это процедура, которая позволяет оценить биоэквивалентность воспроизведенных лекарственных средств без проведения исследований in vivo. Требования к проведению этой процедуры в разных регуляторных документах могут различаться.
ЦЕЛЬ. Сравнение подходов международных и российских регуляторных органов к проведению биовейвера, основанного на биофармацевтической классификационной системе (БКС), рекомендации по проведению теста сравнительной кинетики растворения и определение перспектив дальнейшего совершенствования соответствующих нормативных документов Евразийского экономического союза (ЕАЭС).
ОБСУЖДЕНИЕ. Проанализированы требования к проведению биовейвера, описаны процедуры оценки рН-растворимости и проницаемости лекарственных веществ, выполнения теста сравнительной кинетики растворения в различных средах, моделирующих условия желудочно-кишечного тракта, рассмотрены методики интерпретации полученных результатов. Показана роль вспомогательных веществ, которые оказывают влияние на растворимость и проницаемость действующего вещества.
ВЫВОДЫ. Рекомендован методологический подход к проведению процедуры биовейвера in vitro в качестве замены исследования биоэквивалентности in vivo в соответствии с актуальной нормативной базой ЕАЭС. Перечислены характеристики лекарственных средств, которые являются ограничениями для проведения биовейвера. Обоснована необходимость гармонизации российских и международных требований и руководств.
Волкова Е.А., Медведев Ю.В., Фишер Е.Н., Шохин И.Е. Биовейвер как вид исследования биоэквивалентности. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):42-52. https://doi.org/10.30895/1991-2919-2023-537
Volkova E.A., Medvedev Yu.V., Fisher E.N., Shohin I.E. Biowaiver as a Bioequivalence Study Option. Bulletin of the Scientific Centre for Expert Evaluation of Medicinal Products. Regulatory Research and Medicine Evaluation. 2024;14(1):42-52. (In Russ.) https://doi.org/10.30895/1991-2919-2023-537
В Правилах1, действующих в странах Евразийского экономического союза (ЕАЭС), биовейвер (biowaiver) определяется как процедура оценки биоэквивалентности воспроизведенного лекарственного препарата (ЛП) in vitro без проведения клинических исследований. Использование данной процедуры стало возможным после введения в регуляторную практику биофармацевтической классификационной системы (БКС) в 1995 г. и принятия ее Управлением по контролю за качеством продуктов питания и лекарственных средств (Food and Drug Administration, FDA) [1–3]. Согласно БКС фармацевтические субстанции разделены на 4 группы в зависимости от их биофармацевтической растворимости и проницаемости (табл. 1).
Таблица 1. Характеристики субстанций в соответствии с биофармацевтической классификационной системой (БКС)
Table 1. Active substance characteristics according to the Biopharmaceutics Classification System (BCS)
|
I класс БКС BCS class I |
II класс БКС BCS class II |
|
Высокая биофармацевтическая растворимость Высокая проницаемость (амлодипин [4], вилдаглиптин [5], лакозамид [6, 7] и др.) High biopharmaceutical solubility High permeability (amlodipine [4], vildagliptin [5], lacosamide [6, 7], etc.) |
Низкая биофармацевтическая растворимость Высокая проницаемость (диклофенак [8], ибупрофен [9] и др.) Low biopharmaceutical solubility High permeability (diclofenac [8], ibuprofen [9], etc.) |
|
III класс БКС BCS class III |
IV класс БКС BCS class IV |
|
Высокая биофармацевтическая растворимость Низкая проницаемость (атенолол [10], анастрозол [11], циметидин [12] и др.) High biopharmaceutical solubility Low permeability (atenolol [10], anastrozole [11], cimetidine [12], etc.) |
Низкая биофармацевтическая растворимость Низкая проницаемость (дигоксин [13], ципрофлоксацин [14] и др.) Low biopharmaceutical solubility Low permeability (digoxin [13], ciprofloxacin [14], etc.) |
Таблица составлена авторами / The table is prepared by the authors
При этом следует разделять фармакопейную и pH-растворимость. Согласно требованиям Государственной фармакопеи Российской Федерации (ГФ РФ)2 растворимость субстанции определяют в воде при температуре 20±2 °С, рН-растворимость определяют не менее чем в трех буферных растворах, приготовленных в соответствии с требованиями Фармакопеи ЕАЭС3, при pH 1,2; 4,5 и 6,8 (и, по возможности, при значении, соответствующем величине константы кислотности (pKa), если pKa находится в диапазоне 1,0–6,8) при температуре 37±1 °С [15, 16].
Согласно Правилам4, биовейвер, основанный на БКС, должен включать:
1) экспериментальное изучение рН-растворимости субстанции, использовавшейся для производства воспроизведенного ЛП;
2) получение данных о всасывании (проникающей способности);
3) проведение теста сравнительной кинетики растворения (ТСКР) в трех средах растворения при pH 1,2; 4,5 и 6,8 для 12 образцов каждой серии оригинального (референтного) ЛП и 12 образцов каждой серии воспроизведенного (исследуемого) ЛП.
Международные требования к данной процедуре не противоречат указанным принципам, однако имеют ряд особенностей, например незначительные отличия диапазона рН, критериев приемлемости результатов ТСКР и др. [17]. Кроме того, до 2021 г. согласно руководству FDA5 было рекомендовано изучать рН-растворимость при дополнительных значениях рН, равных pKa±1, однако позже требования были приведены в соответствие с требованиями Международного совета по гармонизации технических требований к лекарственным средствам для медицинского применения (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH) ICH M96.
В Российской Федерации уже имеется достаточный опыт проведения биовейвера [18–20]. Однако переход на регуляторные требования ЕАЭС подразумевает внесение изменений как в процесс выполнения анализа, так и в порядок документирования исследования.
Цель работы — сравнение подходов международных и российских регуляторных органов к проведению биовейвера, основанного на БКС, рекомендации по проведению теста сравнительной кинетики растворения и определение перспектив дальнейшего совершенствования соответствующих нормативных документов ЕАЭС.
Биовейвер допустимо применять при подтверждении биоэквивалентности воспроизведенных лекарственных средств; при внесении изменений в досье, требующих подтверждения фармацевтического подобия; для установления биоэквивалентности как между препаратами, применявшимися в начальных фазах клинических исследований, так и выводимыми на фармацевтический рынок ЕАЭС.
Биовейвер как исследование in vitro взамен исследования биоэквивалентности in vivo допустимо проводить только для препаратов, содержащих субстанции I и III классов БКС [1]. При этом имеется ряд ограничений и требований как к дозированной лекарственной форме препарата, так и к действующему веществу и скорости его высвобождения в среду растворения. Обычно биовейвер проводят для таблеток, капсул и суспензий для приема внутрь с немедленным высвобождением. С рядом ограничений также возможно проведение этой процедуры для таблеток, диспергируемых в полости рта, растворов для приема внутрь, комбинированных ЛП, ректальных лекарственных форм, эмульсий, ЛП липидов для внутривенного парентерального питания, мицеллообразующих препаратов7. Биовейвер нельзя применять к подъязычным, защечным лекарственным формам и лекарственным формам с модифицированным высвобождением. Для возможности проведения процедуры биовейвера в сравниваемых препаратах должны быть одинаковые вспомогательные вещества, способные влиять на биоэквивалентность, в одних и тех же или сопоставимых количествах. Следует отметить, что эти требования в первую очередь относятся к препаратам, содержащим субстанции III класса БКС, а для препаратов, содержащих субстанции I класса, являются рекомендуемыми вследствие меньшего влияния вспомогательных веществ на адсорбцию препарата, однако абсолютно исключить это влияние нельзя. Согласно ICH M98 рекомендуется не превышать 10% различий в составе вспомогательных веществ для препаратов, содержащих субстанции I класса БКС. В документах ЕАЭС указаны более жесткие требования к отклонению содержания вспомогательных веществ в препарате по сравнению с требованиями ICH M9. Сравнение требований приведено в таблице 2.
Таблица 2. Сравнение критериев сопоставимости препаратов по количественному составу вспомогательных веществ
Table 2. Comparison of similarity criteria for medicinal products based on the quantitative excipient composition
|
Вспомогательное вещество, тип или наименование Excipient, type or name |
Допустимое отличие по содержанию в составе препарата (не более), % от общей массы без учета оболочки Acceptable quantitative difference in excipients (maximum), % of the core weight |
|
|
согласно Правилам ЕАЭС12 according to the EAEU requirements12 |
согласно ICH M913 according to ICH M913 |
|
|
Наполнители Fillers |
±5,0 |
±10,0 |
|
Разрыхлители Disintegrants |
||
|
Крахмал Starch |
±3,0 |
±6,0 |
|
Другие разрыхлители Other disintegrants |
±1,0 |
±2,0 |
|
Связующие вещества Binders |
±0,5 |
±1,0 |
|
Смазывающие Lubricants |
||
|
Стеарат магния или кальция Magnesium or calcium stearate |
±0,25 |
±0,5 |
|
Другие смазывающие Other lubricants |
±1,0 |
±2,0 |
|
Скользящие Glidants |
||
|
Тальк Talc |
±1,0 |
±2,0 |
|
Другие скользящие Other glidants |
±0,1 |
±0,2 |
Таблица составлена авторами / The table is prepared by the authors
Именно в оценке влияния вспомогательных веществ на абсорбцию заключается вся сложность выполнения регуляторных требований. Совместная работа технологов и фармакологов по выбору вспомогательного компонента, способного влиять на абсорбцию препарата, позволит принять верное решение о возможности использования процедуры биовейвера. Для доказательства отсутствия влияния вспомогательных веществ на абсорбцию препарата необходимы данные литературы или собственные эмпирические данные. Так, например, несмотря на то что тальк считается веществом, не влияющим на всасывание, он может снижать абсорбцию хинолонов (налидиксовая кислота, циноксацин и др.), фторхинолонов (норфлоксацин, ципрофлоксацин и др.), а также деферипрона, роксадастата и элтромбопага [21–23].
Действующие вещества исследуемого и референтного ЛП также должны быть одинаковыми, допускается использовать субстанции с различием в форме соли только том в случае, если они принадлежат к I классу БКС. Биовейвер запрещен для субстанций сложных эфиров, стереоизомеров и их смесей, комплексов или производных действующего вещества оригинального (референтного) ЛП и веществ с узким терапевтическим диапазоном. И, наконец, биовейвер допустимо проводить только для препаратов, обеспечивающих высвобождение равное или более 85% от номинального содержания действующего вещества в среду растворения в течение 15 или 30 мин, то есть характеризующихся как очень быстро или быстро растворимые9.
В Правилах ЕАЭС и руководстве ICH M9 кратко изложены требования к оценке профиля рН-растворимости, но не приведены примеры ни формата протокола, ни схемы проведения анализа. Более детально подход к определению рН-растворимости описан в руководстве10 Всемирной организации здравоохранения, однако данный документ имеет разночтения с Правилами ЕАЭС. Поэтому разработка собственного протокола проведения испытания рН-растворимости является задачей лаборатории, выполняющей данный вид исследований.
Фармацевтическая субстанция классифицируется как хорошо растворимая, если ее высшая разовая терапевтическая доза полностью растворима в 250 мл или менее буферного раствора11. Согласно руководству ICH M9 рекомендуется
проводить исследование равновесной рН-растворимости методом встряхивания, причем объем среды растворения может быть и менее 250 мл, если имеющееся оборудование позволяет это выполнить. Наиболее оптимальным является использование либо термостатируемого орбитального шейкера для колб вместимостью 5–10 мл, либо термошейкера для микропробирок типа «Эппендорф». Небольшая вместимость микроцентрифужной пробирки позволяет работать с малыми объемами буферного раствора (от 1 до 5 мл) и небольшой навеской субстанции. Масса навески подбирается исходя из информации о растворимости субстанции в указанных средах, либо путем анализа данных литературы (например, DrugBank14 или PubChem15), либо с помощью инструментов с открытым исходным кодом (например, ChemSpider16, Virtual Computational Chemistry Laboratory17 или Swiss ADME18). Необходимо предусмотреть избыток навески, чтобы обеспечить остаток примерно 30–40% нерастворенного твердого вещества. Следует учитывать, что информация о растворимости в литературе и в расчетных программах обычно указывается для температуры 20 °С, а не 37 °С, поэтому требуется значительный избыток навески.
Ни в Фармакопее ЕАЭС19, ни в ГФ РФ20 не описана методика приготовления буферного раствора pH 1,2, поэтому часто вместо буферного раствора используют 0,1 М раствор хлористоводородной кислоты [24, 25]. Такой раствор не обладает буферной емкостью, поэтому при добавлении фармацевтической субстанции может произойти изменение значения рН. Для определения pH-растворимости важно сохранять избыток фармацевтической субстанции, поэтому компенсаторное добавление HCl или NaOH нежелательно, поскольку приведет к растворению излишка твердого вещества. Исходя из собственной практики, использование среды растворения, приготовленной путем смешения растворов NaCl и HCl согласно рекомендациям Фармакопеи ЕАЭС21, для ряда фармацевтических субстанций не решает проблему изменения рН при определении рН-растворимости.
Испытания в каждой из сред растворения проводятся в трех повторностях с выдерживанием образцов в термошейкере при постоянной температуре (37 °С) и перемешивании. Продолжительность инкубации не регламентируется ни Правилами ЕАЭС, ни руководством ICH M9, наиболее рациональным будет индивидуальный для каждого препарата подход к выбору временной точки, сопоставимой с периодом нахождения лекарственной формы в желудочно-кишечном тракте (от 4 до 24 ч). Для определения концентрации вещества в полученном растворе отбирается аликвота надосадочной жидкости и при необходимости разбавляется. Методика количественного определения должна быть валидирована. Высокоэффективная жидкостная хроматография (ВЭЖХ) как метод количественного определения предпочтительнее спектрофотометрии, поскольку позволяет оценивать стабильность образцов и детектировать примеси. Для классификации лекарственного вещества по БКС используется значение самой низкой измеренной растворимости в диапазоне pH 1,0–6,8. Если лекарственное вещество нестабильно с разложением >10% относительно свежеприготовленного образца, то растворимость не может быть определена, и, как следствие, лекарственное вещество не может быть классифицировано22.
Отношение дозы субстанции к растворимости (D/S, мл) рассчитывают по формуле:
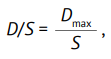 (1)
(1)
где Dmax — максимальная дозировка лекарственного средства, зарегистрированная в Российской Федерации, мг; S — полученное значение растворимости субстанции, мг/мл.
Для субстанций с высокой рН-растворимостью отношение D/S должно быть ≤250 мл.
Для подтверждения степени всасывания (проникающей способности) согласно требованиям Правил ЕАЭС допускается использовать результаты исследований абсолютной биодоступности лекарственного средства или материального баланса на основании обзора литературы. Однако публикуемые статьи могут не содержать необходимых деталей проведенного исследования, позволяющих судить о качестве полученных результатов. Для ряда лекарственных средств, например введенных на рынок десятилетия назад, подобные исследования не проводились.
Дополнительные данные можно получить in silico при помощи, например, инструмента GastroPlus [26] или аналогов [27], однако полученные результаты могут быть использованы исключительно вместе с данными in vivo или in vitro для скрининга и первичного прогнозирования, но не вместо них, поскольку не оценивают важные физиологические параметры, такие как моторика желудка и кишечника, наличие слизи, влияние белков-переносчиков и др. [28]. Применение механистической модели транзита с улучшенной компартментальной абсорбцией (advanced compartmental absorption and transit, ACAT) для имитации всасывания лекарственных средств в желудочно-кишечном тракте при пероральном приеме рассматривается в научной литературе [29, 30]. Согласно руководству ICH M9 в случаях недостатка литературных данных предложено изучать проницаемость валидированными и стандартизированными методами in vitro с использованием клеток Caco-2 (клеточная линия эпителиального рака толстой кишки человека) [31–33]. При культивировании в виде монослоя клетки Caco-2 дифференцируются с образованием плотных контактов между клетками, что служит моделью парацеллюлярного перемещения соединений через монослой. Более того, клетки Caco-2 экспрессируют белки-транспортеры, белки оттока и ферменты конъюгации фазы II для моделирования различных трансцеллюлярных путей, а также метаболической трансформации тестируемых веществ. Во многих отношениях монослой клеток Caco-2 имитирует эпителий кишечника человека [34].
Монослои клеток Caco-2 обычно культивируют на полупроницаемых пластиковых подложках, которые помещают в лунки многолуночных культуральных планшетов. Затем исследуемые образцы с концентрацией, например, 0,01-, 0,1- и 1-кратной от максимальной дозировки, контрольные соединения с известной доказанной проницаемостью (например, ранитидин как образец низкой проницаемости, кетопрофен или пропранолол как образец высокой проницаемости), контроль транспорта гликопротеина-Р (Pgp) (например, родамин 123, дигоксин, паклитаксел, хинидин или винбластин) и ингибитор эффлюкс-транспортера Pgp (например, циклоспорин А) добавляют либо к апикальной (А), либо к базолатеральной (В) сторонам монослоя. Целостность монослоя можно подтверждать измерением сопротивления.
После инкубации из противоположных камер удаляют аликвоты буфера для определения концентрации испытуемых соединений (обычно методом ВЭЖХ с масс-спектрометрическим детектированием) и расчета для каждого соединения показателей проницаемости (называемых коэффициентами кажущейся проницаемости, Papp, см/с) в обоих направлениях (А→В и В→А), а также для определения отношения между ними как разницы в проницаемости, возникающей из-за активного транспорта. Если отношение
Papp В→А/А→В меньше 2,0, то вещество не подвергается активному транспорту и не является субстратом Pgp-транспортера. Если при сравнении кажущейся кишечной проницаемости значение Papp исследуемой субстанции выше соответствующего значения для внутреннего стандарта, например, ранитидина, и ниже, чем Papp пропранолола, это говорит о низкой проницаемости молекулы [33].
Не менее важным требованием для проведения биовейвера, основанного на БКС, является доказательство немедленного высвобождения действующего вещества из ЛП. Проведение теста сравнительной кинетики растворения (ТСКР) требует той же материально-технической базы, что и проведение фармакопейного теста «Растворение». На этом их сходство заканчивается и начинаются принципиальные различия, не позволяющие использовать результаты фармакопейного теста и его валидации для процедуры биовейвера [35, 36].
ТСКР для целей биовейвера проводят на 12 образцах не менее двух серий оригинального (референтного) и не менее двух серий воспроизведенного (исследуемого) ЛП в трех средах растворения (фармакопейные буферы pH 4,5 и 6,8, или имитация кишечного сока без ферментов и 0,1 М раствор HCl pH 1,0–1,2, или имитация желудочного сока без ферментов) при температуре 37±1 °С. Объем среды растворения — не более 900 мл. Для проведения исследования используется либо лопастная мешалка со скоростью вращения обычно 50 об./мин, либо вращающаяся корзинка — обычно 100 об./мин.
Критически важно выбрать верную схему отбора проб, обеспечивающую построение корректного профиля растворения ЛП. Несмотря на общую рекомендацию проводить пробоотбор не реже чем через каждые 15 мин, для препаратов, характеризующихся как быстро растворимые (для которых растворение более 85% действующего вещества составляет менее 30 мин), необходимо сократить интервалы пробоотбора до 5 или 10 мин, особенно в период максимального изменения профиля растворения. Обязательными являются точки 15 и 30 мин, поскольку эти данные позволяют классифицировать препарат как очень быстро или быстро растворимый. Таким образом, оптимальной схемой в рамках биовейера может являться набор точек пробоотбора 5, 10, 15, 20 и 30 мин. Если в течение 15 мин в среду растворения высвободилось более 85% действующего вещества (от номинального количества), то профили растворения признаются эквивалентными без дальнейшей математической оценки. Если в среду растворения высвободилось более 85% действующего вещества в течение 30 мин, то обязательны следующие точки пробоотбора: до истечения 15 мин, на 15-й минуте, точки, когда степень высвобождения составляет около 85% и одна — свыше 85%. Схема 5, 10, 15, 20 и 30 мин удовлетворяет этим требованиям.
Сразу после отбора пробы в качестве превентивной меры возможного сдвига значения рН, изменения объема, концентрации и минимизации отклонений от заданных условий необходимо восполнять отобранный объем среды растворения той же средой и учитывать это в расчетах степени высвобождения23. Следует подчеркнуть необходимость контроля рН, особенно если среда растворения обладает низкой буферной емкостью.
Число серий для выполнения ТСКР оригинального и воспроизведенного ЛП в однозначной трактовке указано в п. 14 приложения 4 к Правилам
ЕАЭС24: «<…> проводить исследование в отношении более чем 1 серии исследуемых лекарственных препаратов». Под исследуемыми препаратами подразумеваются как оригинальный, так и воспроизведенный. Таким образом, ТСКР в рамках биовейера, основанного на БКС, должно проводиться минимум на двух сериях оригинального и на двух сериях воспроизведенного ЛП.
Сопоставимость получаемых профилей растворения обычно оценивают при помощи фактора сходимости (подобия) f2 по формуле:
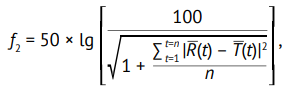 (2)
(2)
где n — количество временных точек; R(t) — среднее значение степени высвобождения действующего вещества из референтного ЛП во временной точке t мин, %; T(t) — среднее значение степени высвобождения действующего вещества из исследуемого ЛП во временной точке t мин, %.
Профили растворения считаются эквивалентными, если значение f2 находится в интервале 50,0–100,0. Результат округляют не до целого, а до первого знака после запятой, поскольку физический смысл f2 заключается в подсчете разницы высвобождения (%) в каждой временной точке, которая должна быть менее 10%. При значении f2<49,9 разница составляет более 10%, при значении f2=49,9 средняя разница в каждый момент времени составляет ровно 10%, при значении f2>50 — менее 10% и при значении f2=100 профили считаются идентичными25. Действительно, если значение |R(t) – T(t)|2/n из формулы (2) составит ровно 100 (10% в квадрате), то корень квадратный (1+100) будет равен 10,04, отношение 100/10,04 равно 9,95, десятичный логарифм 9,95 равен 0,998, таким образом 50×0,998=49,9. При значении f2=49,9 профили признаются неэквивалентными, однако округление результата до целого значения 50 приведет к ложноположительному результату [37].
Для расчета R(t) и T(t) при использовании формулы (2) требуется не менее трех одинаковых временных точек отбора проб для референтного и исследуемого ЛП, не менее 12 результатов определения степени высвобождения действующего вещества (%) для обоих ЛП, не более одного случая превышения уровня 85% для R(t) и T(t), при этом значение относительного стандартного отклонения (RSD) должно составлять не более 20% для первой временной точки и не более 10% для остальных. Полученные профили должны быть одной формы, не иметь S-образных искривлений, не пересекаться друг с другом [37, 38]. Только при выполнении указанных критериев приемлемости допустимо использовать фактор f2 как инструмент оценки сопоставимости профилей растворения. В противном случае используются альтернативные методы, наиболее часто — фактор различия f1, рассчитываемый по формуле [39]:
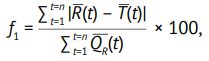 (3)
(3)
где R(t) — среднее значение степени высвобождения действующего вещества из референтного ЛП во временной точке t мин, %; T(t) — среднее значение степени высвобождения действующего вещества из исследуемого ЛП во временной точке t мин, %.
Кроме фактора различия f1 для оценки сопоставимости профилей растворения применимы функция распределения Вейбулла, многомерный анализ, T2-критерий эквивалентности, основанный на расстоянии Махаланобиса, метод бутстреп (bootstrap) для f2, сравнение степеней высвобождения в разных временных точках (например, по t-критерию Стьюдента) и др. [38].
Отчет о проведении ТСКР для целей биовейвера должен содержать: титульную страницу, страницу подписей, краткое описание исследования, перечень сокращений и используемых понятий, информацию о материалах и оборудовании, о сравниваемых ЛП, об аналитическом стандартном образце, условия проведения ТСКР, информацию о маркировке образцов при проведении исследований, описание аналитической методики, результаты анализа исследуемых образцов, перечень используемых программных средств, краткое описание валидации используемой аналитической методики, выводы и заключение, список литературы, приложения и отчет о валидации аналитической методики при проведении ТСКР26.
Методология валидации ТСКР для целей биовейвера требует изложения в рамках отдельной статьи, поскольку биовейвер следует рассматривать как суррогат биоаналитической методики, а не как фармакопейную процедуру.
На основании сравнительного анализа подходов международных и российских регуляторных органов к выполнению основных этапов процедуры биовейвера даны рекомендации по проведению испытаний биофармацевтической (рН-зависимой) растворимости, изучению проницаемости и проведению теста сравнительной кинетики растворения. Введение Правил ЕАЭС в регуляторную практику позволило решить часть вопросов, касающихся проведения биовейвера как испытания in vitro в качестве альтернативы исследованиям биоэквивалентности in vivo. Несмотря на имеющиеся разночтения в тексте Правил ЕАЭС и руководства ICH M9 в отношении рН-растворимости, проницаемости и оценке влияния вспомогательных веществ, сам факт наличия подобного документа — важный шаг для планомерного развития фармацевтической промышленности в ЕАЭС. Следующим логичным шагом будет гармонизация российских и международных требований.
1 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
2 ОФС.1.2.1.0005.15 Растворимость. Государственная фармакопея Российской Федерации. XIV изд. Т. 1. М.; 2018.
3 ОФС 2.2.1.3 Растворы и буферные растворы. Фармакопея Евразийского экономического союза. Т. 1. 2020.
4 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
5 Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. Guidance for industry. U.S. Department of Health and Human Services Food and Drug Administration. Center for Drug Evaluation and Research (CDER); 2017.
6 ICH M9 Biopharmaceutics classification system-based biowaivers. EMA/CHMP/ICH/493213/2018.
7 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
8 ICH M9 Biopharmaceutics classification system-based biowaivers. EMA/CHMP/ICH/493213/2018.
9 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
10 Protocol to conduct equilibrium solubility experiments for the purpose of Biopharmaceutics Classification System-based classification of active pharmaceutical ingredients for biowaiver. Annex 4, WHO Technical Report Series, No. 1019, 2019.
11 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
12 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
13 ICH M9 Biopharmaceutics classification system-based biowaivers. EMA/CHMP/ICH/493213/2018.
14 База данных лекарственных веществ Университета Альберты (University of Alberta) https://go.drugbank.com
15 База данных химических соединений и смесей Национального центра биотехнологической информации США (National Center for Biotechnology Information, NCBI) https://pubchem.ncbi.nlm.nih.gov/
16 www.chemspider.com
17 http://www.vcclab.org
18 База данных Швейцарского института биоинформатики (Swiss Institute of Bioinformatics) http://www.swissadme.ch
19 ОФС 2.2.1.3 Растворы и буферные растворы. Фармакопея Евразийского экономического союза. Т. 1. 2020.
20 ОФС.1.3.0003.15 Буферные растворы. Государственная фармакопея Российской Федерации. XIV изд. Т. 1. М.; 2018.
21 ОФС 2.3.9.1 Рекомендации по проведению испытания на растворение. Фармакопея Евразийского экономического союза. Т. 1. 2020.
22 ICH M9 Biopharmaceutics classification system-based biowaivers. EMA/CHMP/ICH/493213/2018.
23 ОФС.1.4.2.0014.15 Растворение для твердых дозированных лекарственных форм. Государственная фармакопея Российской Федерации. XIV изд. Т. 2. М.; 2018.
24 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
25 EMA’s current practice and challenges in the evaluation of dissolution profile comparisons in support of minor/moderate product quality changes — Case Studies. M-CERSI workshop “In vitro dissolution profiles similarity assessment in support of drug product quality”. Baltimore; 2019. https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/dissolution-similarity/kotzagiorgis-slides.pdf
26 Решение Совета Евразийской экономической комиссии от 03.11.2016 № 85 «Об утверждении Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза».
1. Mishra V, Gupta U, Jain NK. Biowaiver: an alternative to in vivo pharmacokinetic bioequivalence studies. Pharmazie. 2010;65(3):155–61. https://doi.org/10.1691/ph.2010.9231
2. Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. https://doi.org/10.1023/A:1016212804288
3. Becker C, Dressman JB, Amidon GL, Junginger HE, Kopp S, Midha KK, et al. Biowaiver monographs for immediate release solid oral dosage forms: isoniazid. J Pharm Sci. 2007;96(3):522–31. https://doi.org/10.1002/jps.20765
4. Раменская ГВ, Шохин ИЕ, Кулинич ЮИ. Классификации лекарственных веществ по их биофармацевтическим свойствам — БКС и BDDCS. Вестник Воронежского государственного университета. Серия: Химия. Биология. Фармация. 2012;(1):212–5. EDN: OZCXTL
5. Bocci G, Oprea TI, Benet LZ. State of the art and uses for the biopharmaceutics drug disposition classification system (BDDCS): new additions, revisions, and citation references. AAPS J. 2022;24(2):37. https://doi.org/10.1208/s12248-022-00687-0
6. Celso V, Accennato VC, Costa F, Teixeira LS, Abreu F, Mundim IM, et al. Bioequivalence between two tablets of lacosamide in healthy subjects under fasting condition. Int Ann Med. 2018;12(6). https://doi.org/10.24087/IAM.2018.2.6.556
7. Swarnkar G, Joshi P, Shah J, Soni N, Anju G. Solubility determination of lacosamide by HPLC with application to the biopharmaceutics classification system. IJPE. 2011;1(1):51–71.
8. Chuasuwan B, Binjesoh V, Polli JE, Zhang H, Amidon GL, Junginger HE, et al. Biowaiver monographs for immediate release solid oral dosage forms: diclofenac sodium and diclofenac potassium. J Pharm Sci. 2009;98(4):1206–19. https://doi.org/10.1002/jps.21525
9. Potthast H, Dressman JB, Junginger HE, Midha KK, Oeser H, Shah VP, et al. Biowaiver monographs for immediate release solid oral dosage forms: ibuprofen. J Pharm Sci. 2005;94(10):2121–31. https://doi.org/10.1002/jps.20444
10. Vogelpoel H, Welink J, Amidon GL, Junginger HE, Midha KK, Möller H, et al. Biowaiver monographs for immediate release solid oral dosage forms based on biopharmaceutics classification system (BCS) literature data: verapamil hydrochloride, propranolol hydrochloride, and atenolol. J Pharm Sci. 2004;93(8):1945–56. https://doi.org/10.1002/jps.20131
11. Atiyah Altameemi KK, Abd-Alhammid SN. Anastrozole nanoparticles for transdermal delivery through microneedles: preparation and evaluation. J Pharm Negat Results. 2022;13(3):974–80. https://doi.org/10.47750/pnr.2022.13.03.152
12. Jantratid E, Prakongpan S, Dressman JB, Amidon GL, Junginger HE, Midha KK, et al. Biowaiver monographs for immediate release solid oral dosage forms: cimetidine. J Pharm Sci. 2006;95(5):974–84. https://doi.org/10.1002/jps.20614
13. Zakeri-Milani P, Valizadeh H, Tajerzadeh H, Islambulchilar Z. The utility of rat jejunal permeability for biopharmaceutics classification system. Drug Dev Ind Pharm. 2009;35(12):1496–502. https://doi.org/10.3109/03639040903037199
14. Olivera ME, Manzo RH, Junginger HE, Midha KK, Shah VP, Stavchansky S, et al. Biowaiver monographs for immediate release solid oral dosage forms: ciprofloxacin hydrochloride. J Pharm Sci. 2011;100(1):22–33. https://doi.org/10.1002/jps.22259
15. Смехова ИЕ, Перова ЮМ, Кондратьева ИА, Родыгина АН, Турецкова НН. Тест «Растворение» и современные подходы к оценке эквивалентности лекарственных препаратов (обзор). Разработка и регистрация лекарственных средств. 2013;(1):50–61. EDN: QZRXPN
16. Шамаль ЛЛ, Ярушок ТА, Шохин ИЕ, Раменская ГВ. Изучение равновесной биофармацевтической растворимости субстанций ЛС, применяемых при лечении онкологических заболеваний. Ремедиум. 2014;(11):73–6. EDN: TAKAAZ
17. Гребенкин ДЮ, Станишевский ЯМ, Шохин ИЕ. Современные подходы к проведению сравнительного теста кинетики растворения (обзор). Разработка и регистрация лекарственных средств. 2016;(1):166–71. EDN: WBODJF
18. Раменская ГВ, Шохин ИЕ, Савченко АЮ, Кулинич ЮИ, Давыдова КС. Процедура «биовейвер»: современные подходы и общие рекомендации для оценки эквивалентности in vitro лекарственных средств немедленного высвобождения. Рецепт. 2010;(5):33–9. EDN: OIUTUD
19. Шохин ИЕ, Раменская ГВ, Василенко ГФ, Малашенко ЕА. Оценка возможности замены исследований биоэквивалентности in vivo на изучение сравнительной кинетики растворения in vitro (процедура «биовейвер») при определении взаимозаменяемости лекарственных средств («дженериков»). Химико-фармацевтический журнал. 2011;45(2):46–8. EDN: TMQYVH https://doi.org/10.1007/s11094-011-0570-6
20. Шохин ИЕ, Раменская ГВ, Ярушок ТА, Зимакова ЕА, Василенко ГФ, Давыдова КС. Процедура «биовейвер» для витаминных препаратов на примере воспроизведенных лекарственных средств фолиевой кислоты. Вестник Росздравнадзора. 2011;(1):67–71. EDN: QCIMOH
21. Singh GN, Gupta RP. Adsorption characteristics of norfloxacin to pharmaceutical additives. Drug Dev Ind Pharm. 1988;14(13):1845–54. https://doi.org/10.3109/03639048809151991
22. Thapa P, Paudel B. In vitro interactions of ciprofloxacin with selected drugs and excipients. KUSET. 2013;(9):1–14.
23. Бочков ПО, Колыванов ГБ, Литвин АА, Жердев ВП, Шевченко РВ. Влияние высокомолекулярных вспомогательных веществ на оптимизацию фармакокинетических свойств лекарственных препаратов. Фармакокинетика и фармакодинамика. 2016;(1):3–11. EDN: WGCBSZ
24. Дружининская ОВ, Смехова ИЕ. Среды растворения, применяемые в разработке и контроле качества лекарственных средств. Разработка и регистрация лекарственных средств. 2017;(3):144–50. EDN: ZRQDRL
25. Коцур ЮМ, Флисюк ЕВ, Наркевич ИА. Разработка таблеток 4,4’-(пропандиамидо)дибензоата натрия, покрытых кишечнорастворимой оболочкой. Разработка и регистрация лекарственных средств. 2022;11(2):109–17. https://doi.org/10.33380/2305-2066-2022-11-2-109-117
26. Naylor T, Connolly PC, Martini IG, Elder DP, Minekus M, Havenaar R, et al. Use of a Gastro-Intestinal Model and GastroPLUS™ for the prediction of in vivo performance. Industrial Pharm. 2006;12:9–12.
27. Boobis A, Gundert-Remy U, Kremers P, Macheras P, Pelkonen O. In silico prediction of ADME and pharmacokinetics. Report of an expert meeting organised by COST B15. Eur J Pharm Sci. 2002;17(4–5):183–93. https://doi.org/10.1016/s0928-0987(02)00185-9
28. Шохин ИЕ, Раменская ГВ. Методы прогнозирования кишечной проницаемости лекарственных веществ с применением компьютерного моделирования. Биомедицина. 2011;(2):35–40. EDN: NVYEGB
29. Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(1):41–67. https://doi.org/10.1016/s0169-409x(01)00179-x
30. Gigante V, Pauletti GM, Kopp S, Xu M, Gonzalez-Alvarez I, Merino V, et al. Global testing of a consensus solubility assessment to enhance robustness of the WHO biopharmaceutical classification system. ADMET DMPK. 2020;9(1):23–39. https://doi.org/10.5599/admet.850
31. van Breemen RB, Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol. 2005;1(2):175–85. https://doi.org/10.1517/17425255.1.2.175
32. Пятигорская НВ, Кравченко АД. Изучение кишечной проницаемости на модели Caco-2 производного фенилтетрагидрохинолиндиона с TRPA₁-антагонистической активностью. Разработка и регистрация лекарственных средств. 2022;11(3):195–201. https://doi.org/10.33380/2305-2066-2022-11-3-195-201
33. Гребенкин ДЮ, Станишевский ЯМ, Шохин ИЕ, Стойнова АМ, Карпова МА, Корякова АГ и др. Исследование кишечной проницаемости и Pgp-транспорта фосфазида с применением модели Сасо-2. Разработка и регистрация лекарственных средств. 2017;(4):238–42. EDN: ZTWVHZ
34. Шохин ИЕ, Кулинич ЮИ, Раменская ГВ, Кукес ВГ. Применение биологической модели для оценки кишечной проницаемости in vitro — монослоя эпителиальных клеток Сасо-2. Биомедицина. 2012;(3):91–7. EDN: PGJLRV
35. Горбунова ЕВ, Горячев ДВ, Горская ТЕ, Богданов АН. Современные подходы к подтверждению терапевтической эквивалентности лекарственных препаратов локального действия в желудочно-кишечном тракте. Ведомости Научного центра экспертизы средств медицинского применения. 2021;11(4):228–38. https://doi.org/10.30895/1991-2919-2021-11-4-228-238
36. Меньшикова ЛА, Львова АА, Шохин ИЕ, Болдина ЮЕ, Комаров ТН, Медведев ЮВ. Валидация методики количественного определения моксифлоксацина для теста «растворение» методом УФ-спектрофотометрии. Разработка и регистрация лекарственных средств. 2016;(2):94–7. EDN: WYJZKT
37. Yoshida H, Shibata H, Izutsu KI, Goda Y. Comparison of dissolution similarity assessment methods for products with large variations: f2 statistics and model-independent multivariate confidence region procedure for dissolution profiles of multiple oral products. Biol Pharm Bull. 2017;40(5):722–5. https://doi.org/10.1248/bpb.b16-00904
38. Шохин ИЕ, Багаева НС, Малашенко ЕА, Кузина ВН. Методы оценки эквивалентности профилей растворения: современный взгляд (обзор). Разработка и регистрация лекарственных средств. 2020;9(2):145–50. https://doi.org/10.33380/2305-2066-2020-9-2-145-150
39. Сеткина СБ, Хишова ОМ, Зубкевич ЛВ, Каплин АВ, Каплина ЕВ. Влияние биофармацевтических факторов на эквивалентность in vitro воспроизведенных лекарственных средств на основе клопидогреля. Вестник фармации. 2014;(1):33–8. EDN: SEZJSP
Волкова Екатерина Алексеевна, канд. фарм. наук
Трубецкая ул., д. 8, стр. 2, Москва, 119991
Большой б-р, д. 42, стр. 1, Москва, тер. Инновационного центра «Сколково», 121205
Медведев Юрий Владимирович, канд. фарм. наук
Трубецкая ул., д. 8, стр. 2, Москва, 119991
Большой б-р, д. 42, стр. 1, Москва, тер. Инновационного центра «Сколково», 121205
Фишер Елизавета Николаевна, канд. фарм. наук
Трубецкая ул., д. 8, стр. 2, Москва, 119991
Большой б-р, д. 42, стр. 1, Москва, тер. Инновационного центра «Сколково», 121205
Шохин Игорь Евгеньевич, д-р фарм. наук
Научный проезд, д. 20, стр. 3, Москва, 117246
Волкова Е.А., Медведев Ю.В., Фишер Е.Н., Шохин И.Е. Биовейвер как вид исследования биоэквивалентности. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2024;14(1):42-52. https://doi.org/10.30895/1991-2919-2023-537
Volkova E.A., Medvedev Yu.V., Fisher E.N., Shohin I.E. Biowaiver as a Bioequivalence Study Option. Bulletin of the Scientific Centre for Expert Evaluation of Medicinal Products. Regulatory Research and Medicine Evaluation. 2024;14(1):42-52. (In Russ.) https://doi.org/10.30895/1991-2919-2023-537

Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных




























